GO/Fe3O4/有机胺复合材料的制备及对结晶紫染料的吸附性能
2018-08-01亢玺阳杨清香王立杰宋海媚董梦果陈志军
亢玺阳 杨清香 王立杰 宋海媚 张 琰 董梦果 陈志军
(郑州轻工业学院,郑州 450000)
0 引 言
有机染料被广泛用于多个行业的材料着色,如:纺织、染色、造纸和纸浆、制革和油漆等,这些行业的工业废水如果不进行治理会含有过量的染料,而有机染料一般难以降解,并对生物体系造成严重危害而成为水体污染物[1]。制备能够吸附有机染料的吸附剂是治理水体中有机染料污染的重要途径之一,目前吸附剂已成为治理水体污染的研究热点[2-3]。有多种吸附剂被用于水体中染料分子的吸附移除过程,如:活性炭,壳聚糖,改性硅胶、硅藻土、聚合氧化铝、灰烬和天然粘土等[4-6]。但是,这些吸附剂存在吸附量小,吸附染料后的吸附剂需要经过长时间静置沉降才能与水体分离,容易造成二次污染等缺点。因此,研究并开发一种吸附量高并能方便与水体分离的吸附剂,将对水体中染料污染治理具有重要的理论价值和社会经济价值。石墨烯或氧化石墨烯(GO)已被证实可以在某种程度上移除水体中的有机污染物[7],但存在吸附量较小且难以将其与水体分离而导致严重二次污染等缺点[8]。
本文制备了乙二胺 (EDA)功能化的GO/Fe3O4磁性复合材料,乙二胺与丁二胺/己二胺混溶功能化的GO/Fe3O4磁性复合材料,通过胺类有机物对石墨烯的共价键修饰,得到结构更加稳定、比表面积更大且具有超顺磁性的吸附材料。该材料对结晶紫具有良好的吸附性能,且成本低,可简单和快速提取/再生,还可以通过外加磁场实现对吸附剂的快速磁分离从而移除水体中的有机染料[9]。
1 实验部分
1.1 试剂与仪器
浓硫酸购自洛阳市化学试剂厂;石墨粉、高锰酸钾、氢氧化钠、双氧水、三氯化铁、乙酸钠、氯化亚铁、乙二醇、无水乙醇、N,N-二甲基甲酰胺,以上药品试剂均为天津市风船化学试剂有限公司生产;氯仿、硝酸和盐酸购自烟台市双双化工有限公司;乙二胺购自天津市德恩化学试剂有限公司;丁二胺、锂购自阿拉丁试剂有限公司;聚乙烯吡咯烷酮、己二胺购自天津市科密欧化学试剂有限公司;结晶紫购自国药集团化学试剂有限公司。以上试剂均为分析纯,除非特别说明均直接使用。实验用水为超纯水。
样品的TEM测试仪器为河南省表界面重点实验室的高分辨透射电子显微镜 (型号:JEM-2100UHR),加速电压为200 kV。样品的SEM测试是用场发射扫描电子显微镜(型号:JSM-7001F),加速电压为100 kV。使用德国BRUKER公司TENSOR FITR红外光谱仪,漫反射(DRS)测量,样品的结构及骨架振动用KBr做支撑片,在400~4 000 cm-1范围内测量红外吸收峰。使用美国公司Perkin Elmer Diamond TG/DTA热重分析仪进行热分析(样品测试条件:氮气氛围下,加热速率为10℃·min-1,气流速率 200 mL·min-1,温度范围为 30~800 ℃)。 使用德国BRUKER公司AXS D8 X射线衍射仪 (样品测试条件:Cu Kα,λ=0.154 nm,电压 35 kV,电流 20 mA,2θ扫描范围为 5°~90°,步长 0.02°)对制备的样品进行表征。在上海大学测试中心使用高场振动样品磁强计 (LS7307-939)设定在27℃测量磁化强度。使用配备有使用微聚焦Al Kα线(1 486.6 eV)单色化X射线源和半球形电子分析仪的Thermo Scientific Escalab 250Xi光谱仪获得X射线光电子能谱(XPS)。
1.2 制备与测试
1.2.1 氧化石墨烯的制备
参照文献[10-11]制备氧化石墨烯将69 g的浓硫酸(质量分数98%)倒入500 mL三口瓶中,冰浴15 min,将2.0 g石墨粉缓慢加入到浓硫酸中,冰浴30 min,继续缓慢加入8.0 g高锰酸钾,加完后将装置放入35℃水浴中,搅拌12 h。搅拌后缓慢加6次水,每次46 mL,在35℃条件下搅拌2 h。然后缓慢加入25 mL 30%的H2O2继续搅拌15 min,反应暂停。把黄色溶液倒进离心管中用离心机离心5 min,转速8 000 r·min-1。将得到的黄色粘稠物用稀盐酸(150 mL盐酸与900 mL水配制)洗涤6次,最后将流动性较强的黄色粘稠物均匀涂在玻璃板上,70℃下烘干。
注意:加高锰酸钾必须缓慢!用时1~2 h。
1.2.2 乙二胺修饰氧化石墨烯
量取 400 mL的乙二胺,加入到1 000 mL三口烧瓶中。在氮气氛围中加350 mg的锂(碎块),待锂完全溶解后,升高温度(<30℃)使溶液逐渐变为深蓝色,搅拌至溶液由深蓝色变为灰白色。向反应体系中加入400 mg自制的GO,升温至50℃搅拌12 h。加入75 mL甲醇和250 mL水,将反应悬浮液过滤,乙醇和水各洗3次,60℃下真空干燥12 h。得到乙二胺功能化的GO[12]。
实验过程中发现如果先加锂,使锂在乙二胺中溶解,在升温的过程中溶液的颜色由深蓝色变为灰白色,然后加石墨烯,可以得到稳定的石墨烯悬浮液。造成上述现象的原因是锂首先与乙二胺发生反应,生成单取代的锂胺衍生物作为亲核试剂进攻石墨烯的C=C,生成乙二胺功能化的石墨烯。为了消除石墨烯物理吸附乙二胺的影响,用过量的醇和蒸馏水反复洗产物。
1.2.3 乙二胺和丁二胺混溶修饰氧化石墨烯
先将丁二胺(m.p.27~28℃)熔化为液体。用量筒量取120 mL的乙二胺和30 mL的丁二胺,加入到500 mL的三口烧瓶中。在氮气氛围中加130 mg的锂,待锂完全溶解后,升高温度(<30℃)使溶液逐渐变为深蓝色。搅拌至溶液由深蓝色逐渐变为灰白色。向反应体系中加入150 mg自制的GO,升温至 50℃,搅拌12 h,之后加28.2 mL甲醇和 93.8 mL水。将反应悬浮液过滤,乙醇和水各洗3次,60℃下真空干燥12 h。得到乙二胺和丁二胺混溶功能化的GO。 依次再制备 V乙二胺∶V丁二胺=2∶1、3∶1、5∶1、6∶1 的混溶功能化GO。
1.2.4 乙二胺和己二胺混溶修饰氧化石墨烯
先将己二胺(m.p.39~43℃)熔化为液体。量取160 mL的乙二胺和40 mL的丁二胺,加入到500 mL三口烧瓶中。在氮气氛围中加200 mg的锂,待锂完全溶解后,升高温度(<30℃)使溶液逐渐变为深蓝色,搅拌至溶液由深蓝色逐渐变为灰白色。向反应体系中加入200 mg自制的GO,升温至50℃,搅拌12 h。之后加37.5 mL甲醇和125 mL水。将反应悬浮液过滤,乙醇和水各洗3次,60℃下真空干燥12 h。得到乙二胺和己二胺混溶功能化的GO。依次制备V乙二胺∶V己二胺=2∶1、3∶1、5∶1、6∶1 的混溶功能化 GO。
1.2.5 四氧化三铁的制备
称量 1.800 3 g FeCl3·6H2O,2.002 5 g 乙酸钠,1.006 1 g聚乙烯吡咯烷酮(PVP)溶解在50 mL乙二醇中,搅拌成均匀透明澄清液体。然后将液体转移至高压釜中,200℃下反应8 h。倒出反应釜中的液体,磁铁分离,乙醇洗3次,60℃下真空干燥[13-15]。
1.2.6 复合物的制备
称量0.50 g氨基化GO分散在40 mL二甲基甲酰胺(DMF)中,称量0.40 g Fe3O4分散在60 mL氯仿中,搅拌、超声使其形成均匀分散液。4 h后将复合物用无水甲醇洗涤3次,磁铁分离,60℃下真空干燥。将上述的复合物进行编号:乙二胺功能化的GO/Fe3O4磁性复合材料(E),乙二胺和丁二胺混溶功能化的 GO/Fe3O4磁性复合材料(体积比为 2∶1、3∶1、4∶1、5∶1 和 6∶1 分别标记为 B1、B2、B3、B4 和 B5),乙二胺与己二胺混溶功能化的GO/Fe3O4磁性复合材料(体积比为 2∶1、3∶1、4∶1、5∶1 和 6∶1 分别标记为 H1、H2、H3、H4 和 H5)。
1.2.7 时间对吸附染料质量浓度的影响
称量50 mg结晶紫置入100 mL棕色容量瓶中,定容、超声。将制备的有机胺功能化的GO/Fe3O4复合材料各称量20 mg,倒入50 mL离心管中,将配制好的结晶紫染料取25 mL倒入离心管中,恒温振荡。每隔1 h取一次液体,磁铁分离,取上层澄清液体用紫外光谱测试吸附染料的质量浓度。
1.2.8 染料的质量浓度对吸附染料浓度的影响
选取编号为E、B4和H4这3个样品各称量10 mg放入50 mL离心管中。配制质量浓度为100、200、300、400、500 mg·L-1的结晶紫染料, 取 10 mL入离心管中,恒温振荡。过夜,磁铁分离,取上层澄清液体用紫外光谱测试吸附染料的质量浓度。
2 结果与讨论
2.1 TEM和SEM分析
图1 GO、Fe3O4和复合材料E的TEM图。从图1a可以看出,GO是片状的,具有多层结构,表面光滑,片层明显起皱。层与层之间的相互折叠和GO极薄的平面结构产生较大的比表面积,这有利于提高吸附能力。由于GO的长度相对较长并且具有仅几纳米薄的厚度,所以石墨烯纳米薄片在溶剂中表现出最低的能量并彼此折叠。在TEM照片中可以清楚的看到,石墨烯纳米片具有大的对比度的暗线,应是折叠的多层石墨烯氧化物。基于分析,制备的样品具有GO的形态特征。从图1b看出,Fe3O4的形状是球形的,具有均匀的粒度,平均粒径约为200 nm。从图1c和1d可以看出,复合材料E中的Fe3O4很好地固定在样品中薄薄的GO片层上。GO片层不仅可以防止Fe3O4微球的团聚,而且可以很好地分散这些氧化物微球,并且还可以显著增强复合材料的比表面积,提高复合材料的吸附性能。

图1 GO(a)、Fe3O4(b)和复合材料 E(c~d)的 TEM 图Fig.1 TEM images of GO(a),Fe3O4(b)and composites E(c~d)
图2 为复合材料E、B4和H4的SEM图。从图2中可以看出,复合材料H4(图2e,2f)要比复合材料E(图2a,2b)、B4(图2c,2d)更好地分隔 GO 片层的结构,可以把Fe3O4很好地包裹起来,而Fe3O4也很好地穿插在GO片层上。这是由于己二胺的分子链长比丁二胺的长,而丁二胺的又比乙二胺的长,所以H4中的己二胺可使GO的结构更易分层。另外,由于Fe3O4具有磁性,所以图中的Fe3O4有团聚现象。

图2 复合材料 E(a~b)、B4(c~d)和 H4(e~f)的 SEM 图Fig.2 SEM images of composites E(a~b),B4(c~d)and H4(e~f)
2.2 红外光谱图
复合材料的FT-IR光谱如图3所示。在557 cm-1处产生的强吸收带是Fe-O键,可以看出Fe3O4成功附着在氨基化GO表面。在3 433 cm-1处产生的强而宽的吸收峰是-NH2的伸缩振动。在1 076 cm-1处对应于C-O键的伸缩振动,在1 400 cm-1处附近则是-OH的面内变形振动峰,峰的强度较弱,峰形较宽。在1 629 cm-1处为石墨中sp2杂化C=C双键伸缩振动[16],而2 926 cm-1处则为C-H吸收峰不对称伸缩振动。红外光谱数据表明,所制样品为有机胺功能化的GO/Fe3O4复合材料。
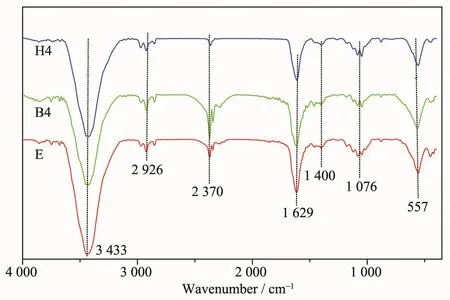
图3 复合材料E、B4和H4的红外谱图Fig.3 FT-IR spectra of composites E,B4 and H4
2.3 热重分析(TG)
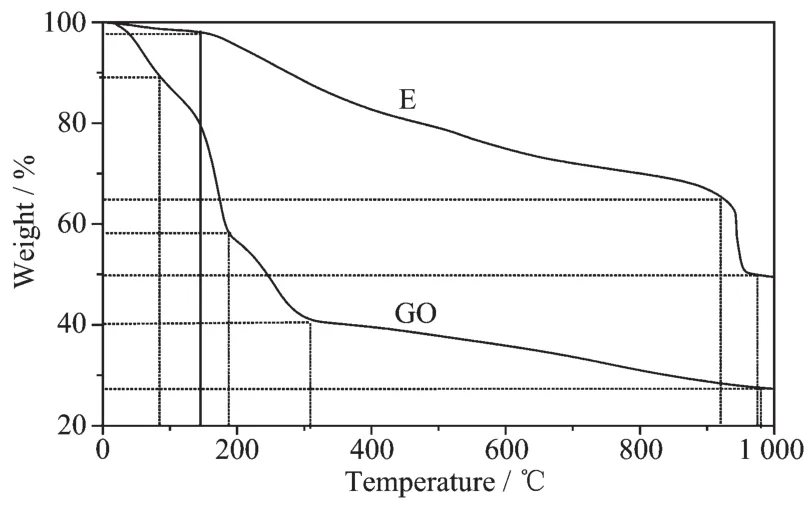
图4 GO与复合材料E的TG曲线Fig.4 TG curves of GO and composites E
GO与复合材料E的热重曲线如图4所示(复合材料B4和H4的失重与E大致相同,故未列出)。可以看出,GO与复合材料E的失重大致分为3个阶段。第一阶段,GO在20~187℃大约有17.2%的失重,而复合材料E在20~145℃大约有2.2%的失重,对应于GO中存在有的少量水和乙醇 (沸点为78.3℃)等,包括物理吸附和化学键合水,表明GO有很强的亲水性。第二阶段,GO在130~306℃大约有42.2%的失重,是由于GO表面上的含氧官能团(如-OH,-COOH,-O-和C=O)受热分解导致。而复合材料在145~922℃大约有32.6%的失重,这是GO表面上的含氧官能团和C-C键碳骨架在依次受热分解。第三阶段,GO在306~983℃失重大约有13.0%,这就是GO上C-C键碳骨架在受热分解。复合物在922~983℃大约有15.4%的失重,这可能是氨基在受热分解。复合物在1 000℃的时候质量分数要比氧化石墨烯的质量分数多出约22.2%,这应该是Fe3O4(熔点为 1 594 ℃)。
2.4 X射线衍射(XRD)图和磁滞回线
如图5a所示,复合材料E的衍射峰出现在18.47°,30.32°,35.67°,43.24°,53.62°,57.16°,62.82°,分别对应于立方相 Fe3O4的(111),(220),(311),(400),(422),(511),(440)晶面,与标准衍射卡 PDF#19-0629上的衍射峰一致[15]。GO在2θ=10.16°处产生了尖锐的衍射峰,对应于晶面(001)[17]。而有机胺功能化的GO/Fe3O4复合材料B4、H4的衍射峰大致与Fe3O4的相同,GO的衍射峰不太明显。有机胺的加入未使复合物产生相变。测定的复合材料E的磁滞回线如图5b所示。其磁滞回线为S型曲线,故E是超顺磁性的,比饱和磁化强度 Ms为 53.0 emu·g-1。
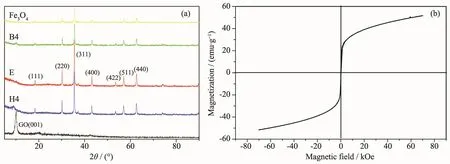
图5 (a)GO、复合材料E,B4、H4以及Fe3O4的XRD图;(b)复合材料E的磁滞回线图Fig.5 (a)XRD patterns of GO,composites E,B4,H4 and Fe3O4;(b)VSM of composites E
2.5 X射线光电子能谱(XPS)分析
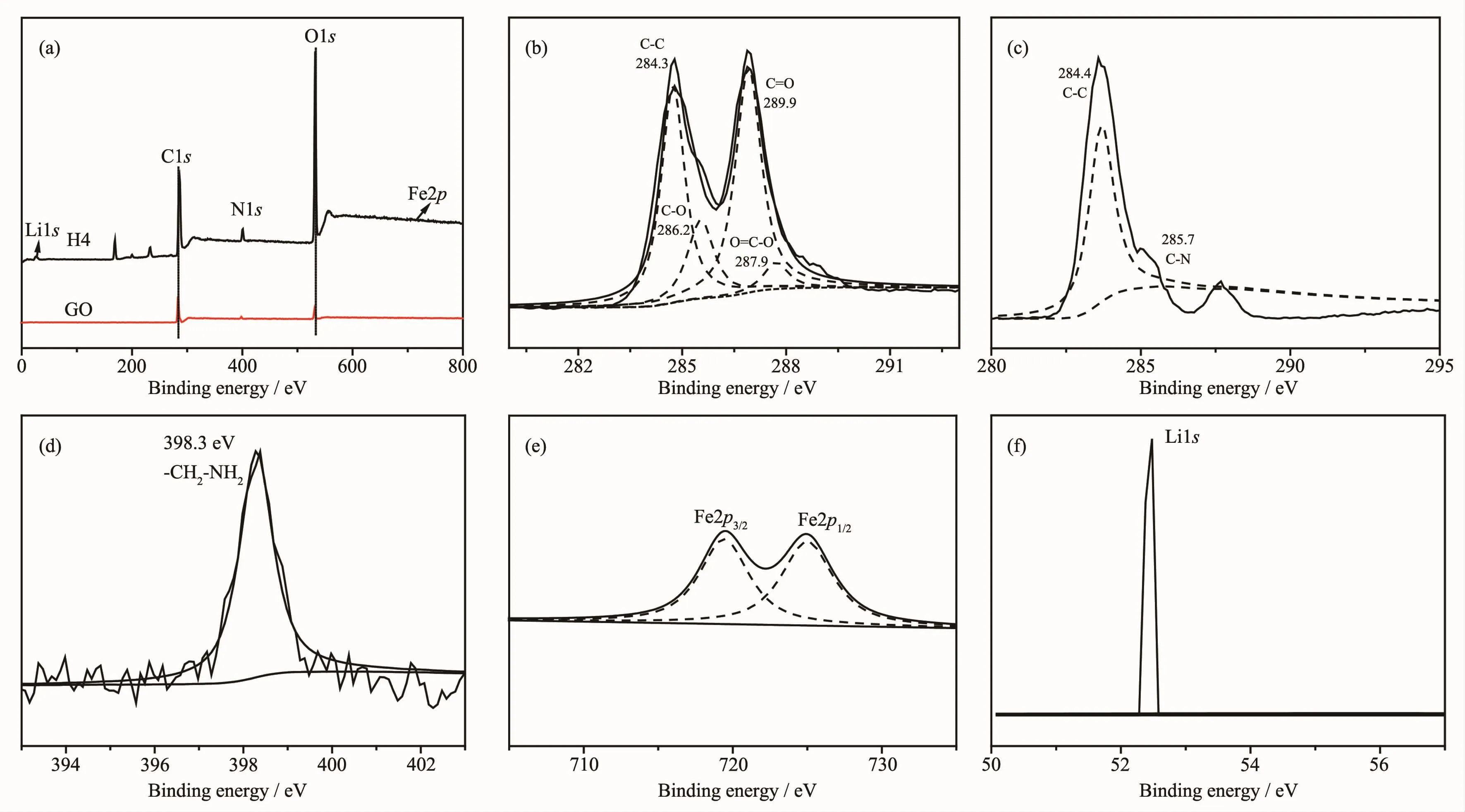
图6 GO 和 H4(a),GO C1s(b),H4 C1s(c),H4 N1s(d),H4 Fe2p(e)和 Li1s(f)的 XPS 谱Fig.6 XPS spectra of GO and H4(a),GO C1s(b),H4 C1s(c),H4 N1s(d),H4 Fe2p(e)and Li1s(f)
XPS分析可用于确定改性所引入的化学物质。图6a中,在285.6、400.6和531.9 eV处的峰分别对应于 C、N 和 O[18],在 52.5 eV 处的峰为 Li[19],位于719.4 eV峰为Fe[20]。图6b中,位于284.4 eV的GO的C1s峰其结合能位于284.3、286.2、286.9和 288.4 eV,它们分别对应 C-C,C-O,C=O 和 O=C-O[20]。 图6c描述了H4的C1s,其中C-C键的主峰位于284.4 eV,胺(CH2-NH2)中的C-N键的第二个峰位于285.7 eV[21]。从图6d可以看出,N1s结合能位于398.4 eV,对应于-CH2-NH2[22]。图6e证明了Fe的组成[20,23]。图6f中位于52.5 eV的结合能为Li1s[19]。
2.6 时间对吸附染料质量浓度的影响
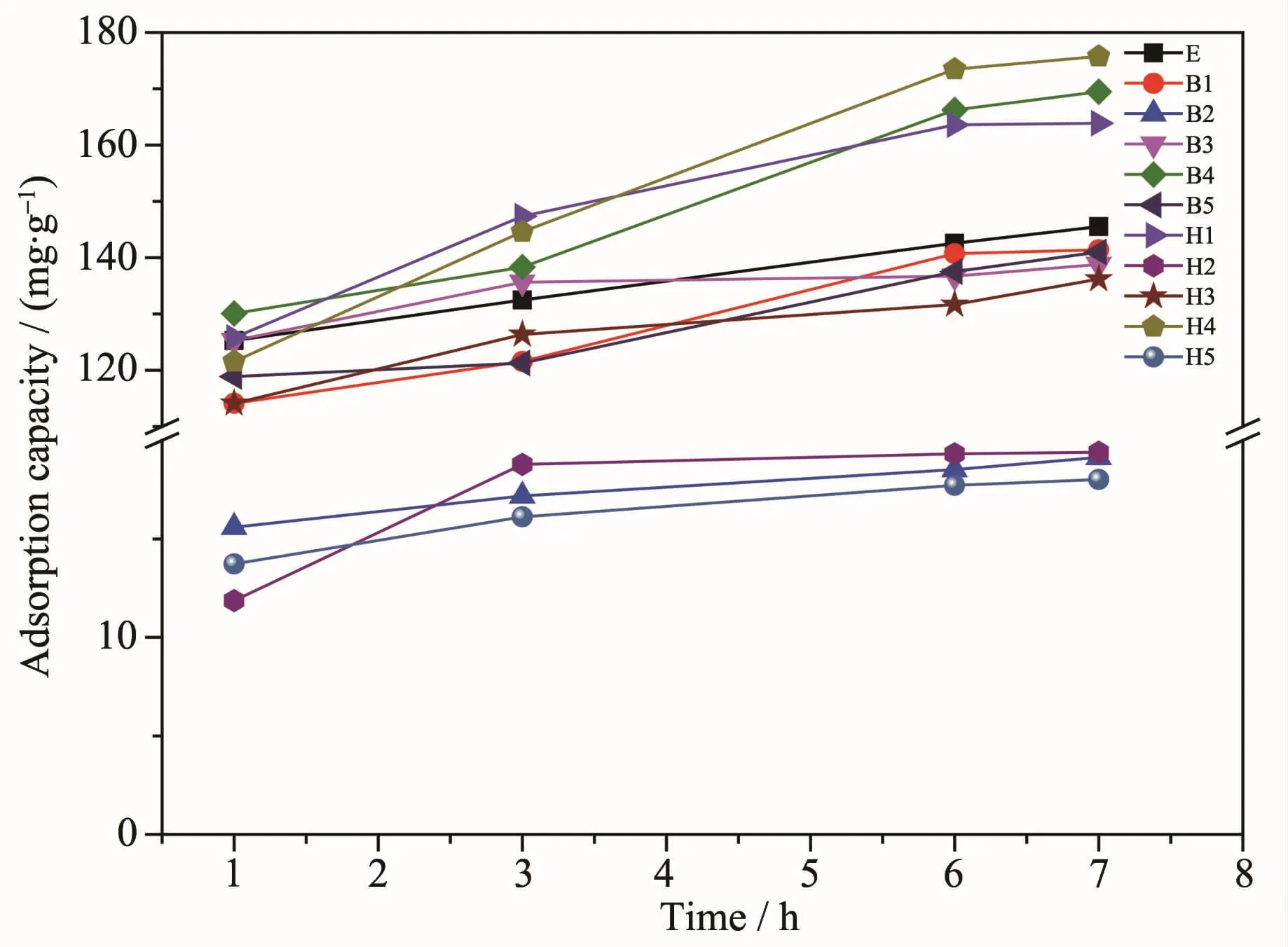
图7 不同复合材料对结晶紫最大吸附量随时间的变化Fig.7 Change of maximum adsorption capacity of crystal violet on different composites with time
图7 表示不同药品对结晶紫染料最大吸附量随时间的增加而增加,结晶紫染料浓度为500 mg·L-1。从图7中可以看出,有机胺功能化的GO/Fe3O4磁性复合材料在吸附结晶紫染料时,随着时间的增加,复合材料对结晶紫的吸附量越大。由实验可得6 h后最大吸附量基本不再变化。从图7中可以看出,乙二胺与己二胺混溶功能化的GO/Fe3O4磁性复合材料H4对结晶紫染料的吸附量最大,为173.2 mg·g-1。
2.7 染料浓度对吸附染料质量浓度的影响
根据时间对吸附结晶紫染料的影响,选取复合材料E、B4和H4。从图8中可以看出,复合材料对结晶紫染料的最大吸附量随质量浓度增大而增大,而移除率却随结晶紫染料质量浓度增大而减小,最后都趋于定值。由这3种复合材料比较得出复合材料H4吸附结晶紫染料的量最多,B4其次,这验证了己二胺要比乙二胺和丁二胺的分子链要长,它改性GO要比乙二胺和丁二胺这2种药品对GO改性分层隔离得要开,比表面积变大吸附性能增强。结晶紫染料的移除率可表示为:
E=(C0-Ce)/C0×100%
其中,E为移除率,C0和Ce分别为结晶紫染料的初始质量浓度和吸附处理后的质量浓度。复合材料H4在浓度为 400 mg·L-1时最大吸附量为 164.3 mg·L-1,浓度为100 mg·L-1时最大移除率为68.1%。
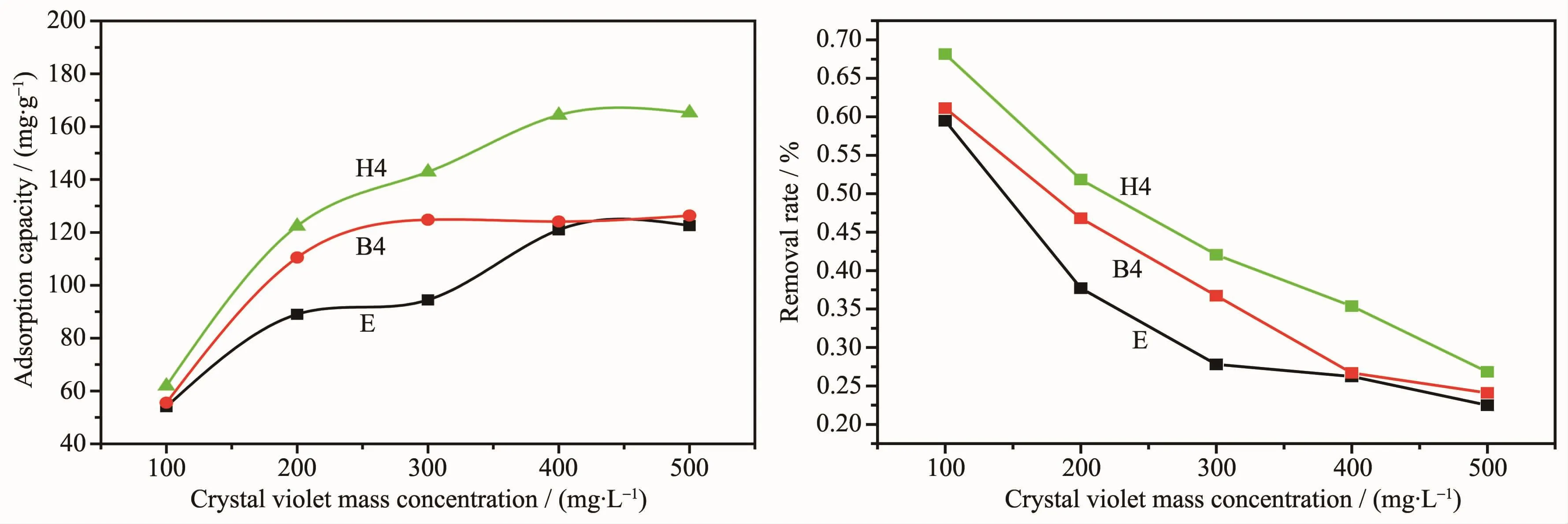
图8 复合材料E、B4、H4吸附结晶紫的最大吸附量和移除率Fig.8 Maximum adsorption amount and the removal rate of the crystal violet on composites E,B4 and H4
3 结 论
用乙二胺、乙二胺与丁二胺/己二胺混溶来改性氧化石墨烯,并用物理混合法制备了GO/Fe3O4/有机胺的三元复合体系。复合物中,GO为片层结构,Fe3O4粒径分布均匀,可以很好地穿插在氨基化GO片层上,由于己二胺的链长更长,它要比乙二胺和丁二胺更好地隔离GO片层。
磁性复合材料对结晶紫染料具有吸附能力,且能够借助Fe3O4的磁性,实现快速分离。乙二胺和己二胺混溶功能化的GO/Fe3O4磁性复合材料要比其它2种胺改性的复合材料吸附更好,对结晶紫的最大吸附量随染料的浓度增大而增大,随时间增长而增大,最后都趋于定值。 其中,V乙二胺∶V己二胺=5∶1 时制得的复合材料H4吸附性能最好。
