同位素稀释-气相色谱-串联质谱法测定化妆品中10种合成麝香
2018-07-27张晓岚徐红斌
周 静,张晓岚,徐红斌
(1.上海市质量监督检验技术研究院,上海 200233;2.上海大学环境与化学工程学院,环境污染与健康研究所,上海 200444)
麝香是化妆品的重要原料之一,被广泛用于香水、花露水、剃须水、膏霜等产品中。由于天然麝香的资源有限,19世纪末,Baur等[1]首次合成了硝基麝香。合成麝香香味独特、定香能力好,逐渐成为香精香料行业中天然麝香的廉价替代物。合成麝香主要分为硝基麝香、多环麝香和大环麝香3类。由于大环麝香的制造工艺复杂、成本高,在香精香料行业中应用很少,而硝基麝香和多环麝香大约占了该行业中95%以上的市场份额。合成麝香具有较强的亲脂憎水性,在环境中难降解,易生物富集,对生物和人体均呈现一定的生物毒性,易渗入人体细胞且具有疑似的致癌性[2-4]。因此,美国及欧盟的许多国家已将其列为高关注物质,并禁止或限制将其用于化妆品及与皮肤接触的产品中[5-7]。我国2015年修订的《化妆品安全技术规范》[8]中明确:将伞花麝香、葵子麝香和西藏麝香列为禁用物质,另规定了酮麝香的使用限量为香水1.4%、淡香水0.56%、其他0.042%;二甲苯麝香的限量为香水1.0%、淡香水0.4%、其他0.03%。GB/T 22731—2017《日化香精》[9]中除了上述禁用的3种麝香外,将二甲苯麝香和万山麝香也列在禁用名单中。然而,目前还没有日化产品中合成麝香的标准检测方法,给行政执法及进出口贸易带来不便。因此,建立准确、快速、灵敏的化妆品中合成麝香的分析方法十分必要。
目前,文献报道的化妆品中合成麝香分析方法的研究主要集中于禁、限用的硝基麝香。测定方法多以气相色谱-质谱(GC/MS)法[10-12]和气相色谱-串联质谱(GC-MS/MS)法[13-16]为主。质谱法可以弥补色谱法无法分离复杂组分和假阳性的缺点,同时缩短合成麝香的分离时间。与单级质谱相比,串联质谱的多反应监测(MRM)模式具有更强的抗基质干扰能力和定性、定量准确性,可以实现对化妆品中合成麝香的准确测定。马强等[13]报道了固相萃取-同位素稀释-气相色谱-串联质谱法测定化妆品中的二甲苯麝香;王征[14]和郑小严[15]采用GC-MS/MS法测定了化妆品中5种硝基麝香类化合物;叶洪等[16]应用GC-MS/MS法测定了个人护理品中9种合成麝香;Dong等[17]采用载体辅助液液萃取和固相萃取的前处理技术,结合GC-MS/MS法实现了膏霜中7种人工合成麝香的同时测定。还有一些研究者运用GC-MS/MS开发了水产品和空气等样品中多种合成麝香的同时检测方法[18-20]。然而,以上检测方法存在前处理复杂或样品基质单一等问题。如叶洪等[16]开发的检测方法仅适用于测定沐浴露、洗发露、洗衣液等液体类样品,且采用荧蒽-D10作为内标物,与麝香类物质的理化性质存在一定差异,无法真实地反映方法的有效性与准确性。Dong等[17]的研究仅涉及膏霜类样品,且前处理相对繁杂,不利于方法的标准化推广。
本研究拟采用麝香类同位素标样稀释,结合超声波有机溶剂萃取和气相色谱-串联质谱法检测,在文献[21]的前处理方法基础上深入优化,充分利用串联质谱强大的抗基体干扰能力,省去前处理中基体分散固相法净化步骤,希望建立化妆品中两类(硝基麝香和多环麝香)共10种合成麝香同时检测的分析方法,并通过对市售几类化妆品样品的检测论证该方法的可行性。
1 实验部分
1.1 主要仪器与装置
7890A-7000B气相色谱-三重四极杆串联质谱联用仪(配有电子轰击源(EI)和MassHunter工作站),VF-WAXms毛细管色谱柱(30 m×0.25 mm×0.25 μm):美国Agilent公司产品;KUDOS型超声仪:科岛超声仪器有限公司产品;TB-114电子天平,3-30K型高速冷冻离心机:北京塞多利斯仪器系统有限公司产品。
1.2 主要材料与试剂
正己烷、乙酸乙酯、乙腈、二氯甲烷、丙酮和甲醇:色谱纯,美国Thermo Fisher公司产品;氯化钠:分析纯,国药集团化学试剂有限公司产品。
佳乐麝香(Galaxolide, HHCB)、吐纳麝香(Tonalide, AHTN)、粉檀麝香(Phantolide, AHMI)、开许梅陇(Cashmeran, DPMI)、特拉斯(Traseolide, ATII)、西藏麝香(musk tibetene, MT)、萨利麝香(Celestolide, ADBI)、二甲苯麝香(Musk xylene, MX)和酮麝香(Musk ketone, MK)等标准品(纯度均大于95%),氘代吐纳麝香(d3-AHTN)和氘代二甲苯麝香(d15-MX)(纯度大于98%):德国Dr. Ehrenstorfer公司产品;葵子麝香(Musk ambrette, MA):纯度大于98%,美国AccuStandard公司产品。
标准储备液的配制:分别称取0.1 g(精确至0.000 1 g)10种合成麝香标准品于100 mL容量瓶中,用正己烷溶解并定容,得到1.0 g/L合成麝香标准储备液。以同样的方法分别得到1.0 g/L的两种氘代内标(d3-AHTN和d15-MX)标准储备液。
混合标准中间工作液的配制:用正己烷将10种合成麝香和2种氘代内标的标准储备液分别稀释成1.0 mg/L的混合标准中间工作液。
标准工作液的配制:分别准确移取1、2、5、10、20、50、100 μL的1.0 mg/L的10种合成麝香混合标准中间工作液,各加入50 μL 1.0 mg/L氘代内标混合标准中间工作液,用正己烷定容至1.0 mL,配制成质量浓度为1.0、2.0、5.0、10.0、20.0、50.0、100.0 μg/L的系列混合标准工作液,内标浓度均为50.0 μg/L,于4 ℃保存。
1.3 实验条件
1.3.1样品前处理 称取约1.00 g样品于15 mL具塞刻度离心管中,加入50 μL 1.0 mg/L的d3-AHTN和d15-MX氘代混标溶液,再加入1.0 mL饱和氯化钠水溶液和2.0 mL正己烷提取剂,于40 ℃超声提取20 min,以8 000 r/min离心3 min,取上清液,剩余样品残渣用正己烷(每次2.0 mL)反复提取2次,合并上清液,氮吹浓缩至1.0 mL,过0.45 μm滤膜,待GC-MS/MS检测。
1.3.2色谱条件 色谱柱:VF-WAXms 石英毛细管柱(30 m×0.25 mm×0.25 μm);升温程序:初始温度80 ℃,以10 ℃/min升至160 ℃,再以5 ℃/min升至175 ℃,保持5 min,以1 ℃/min 升至178 ℃,保持1 min,以20 ℃/min升至220 ℃,保持8 min;载气(He)流速1.0 mL/min;进样量1.0 μL;进样方式:不分流进样,0.75 min后开阀。
1.3.3质谱条件 电子轰击(EI)离子源;电子能量70 eV;传输线温度250 ℃;离子源温度150 ℃;四极杆温度230 ℃;质谱仪接口温度250 ℃;离子监测模式:多反应监测模式(MRM)。监测离子对与碰撞能量等信息列于表1。
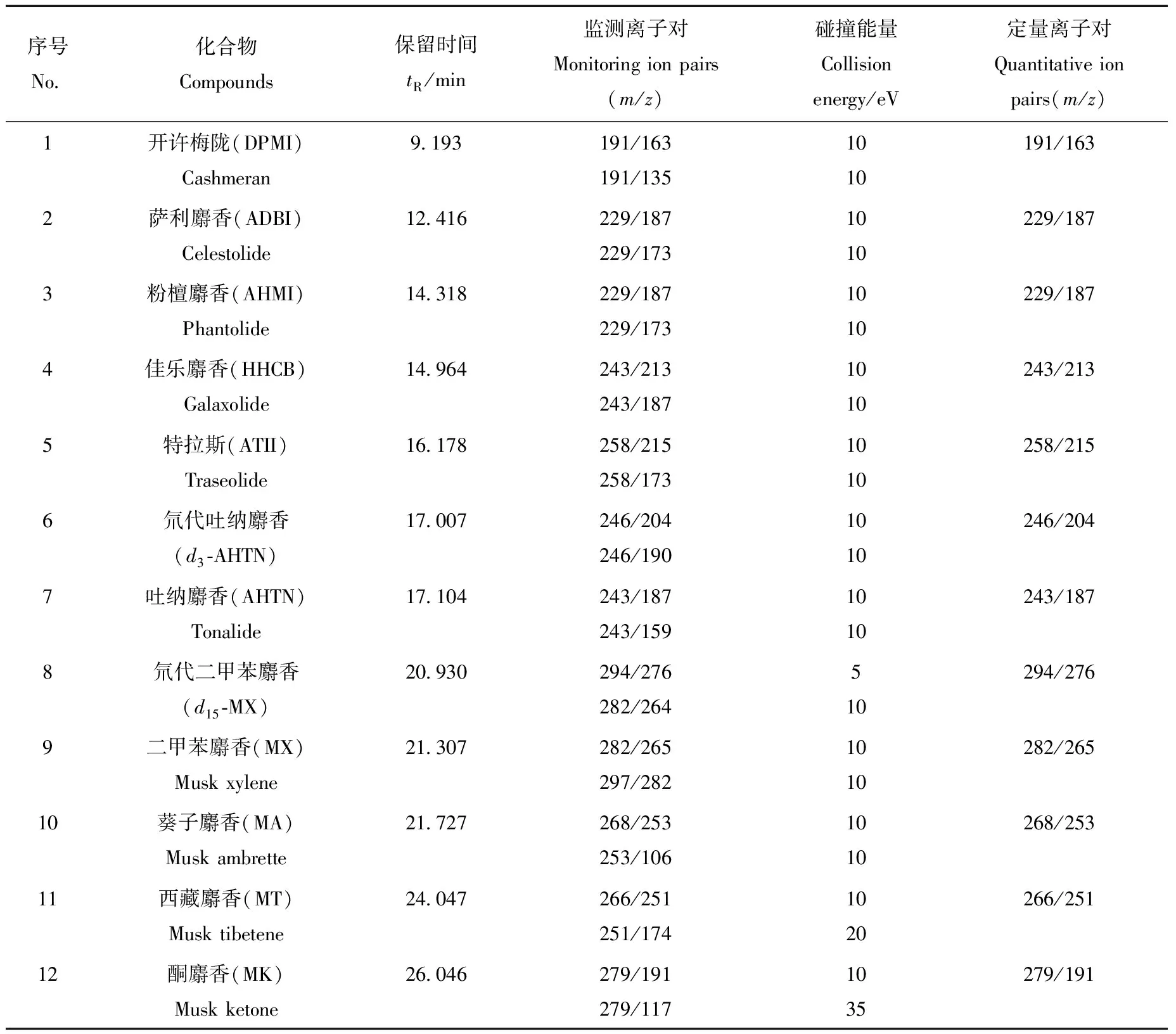
表1 10种合成麝香和2种氘代内标的串联质谱分析参数Table 1 Analysis parameters of mass spectrometry for 10 synthetic musks and 2 deuterated internal standard
2 结果与讨论
2.1 前处理方法的优化
2.1.1内标物质的选择 在样品前处理过程中,选取与目标物质结构一致的稳定同位素内标,能够有效减少提取及净化过程中的损失对检测结果的影响。在质谱检测中,以稳定同位素内标进行定量校正,能够消减不同基质对待测物响应的抑制或增强作用,提高定量的准确性。d3-AHTN和d15-MX的物理及化学性质基本与目标分析物一致,且在化妆品中不存在该类化合物,因此适合作为测定化妆品中合成麝香含量的内标物质。本方法选用d3-AHTN作为6种多环麝香(DPMI、ADBI、AHMI、HHCB、ATII和AHTN)的内标物质,选择d15-MX作为4种硝基麝香(MX、MA、MT和MK)的内标物质。
2.1.2提取溶剂的优化 由于乳液、膏霜等化妆品的乳化体系分水包油和油包水两种类型,其中的待测物有可能被包于化妆品乳化颗粒最内层,如果直接采用溶剂提取,样品分散性不好,会限制内层待测物的提取率,造成提取效率偏低。故本方法先采用1.0 mL饱和氯化钠水溶液将样品充分分散,再用有机溶剂分次提取,以达到更好的提取目的。另外,饱和氯化钠水溶液的加入可产生盐析作用,减少样品乳化效应的产生。
合成麝香极性较小,具有脂溶性,难溶于水,在弱极性或非极性溶剂中有较好的溶解度。根据相似相溶原理,并考虑到提取溶剂的普适性,选择了正己烷、乙酸乙酯、乙腈、二氯甲烷、丙酮和甲醇作为提取溶剂,考察各提取溶剂对膏霜类化妆品中合成麝香的提取效率。具体操作如下:称取1.00 g空白膏霜样品,加入10 μL 1.0 mg/L的10种麝香混合标准工作中间液(即样品加标水平10.0 μg/kg)混匀,再加入提取溶剂和1.0 mL饱和氯化钠水溶液,后续操作参照1.3.1节。每种提取溶剂重复6次实验,提取效率以每种合成麝香的6次平均绝对回收率计算。结果表明,甲醇与饱和氯化钠溶液体系互溶,无法分层;乙腈、丙酮和二氯甲烷存在部分互溶,且都有不同程度的乳化现象;正己烷和乙酸乙酯对10种合成麝香的提取效率最高,且正己烷提取溶剂的干扰和杂质峰较乙酸乙酯的少,因而选择正己烷作为提取溶剂。使用不同提取溶剂时,膏霜类化妆品中10种合成麝香的回收率列于表2。
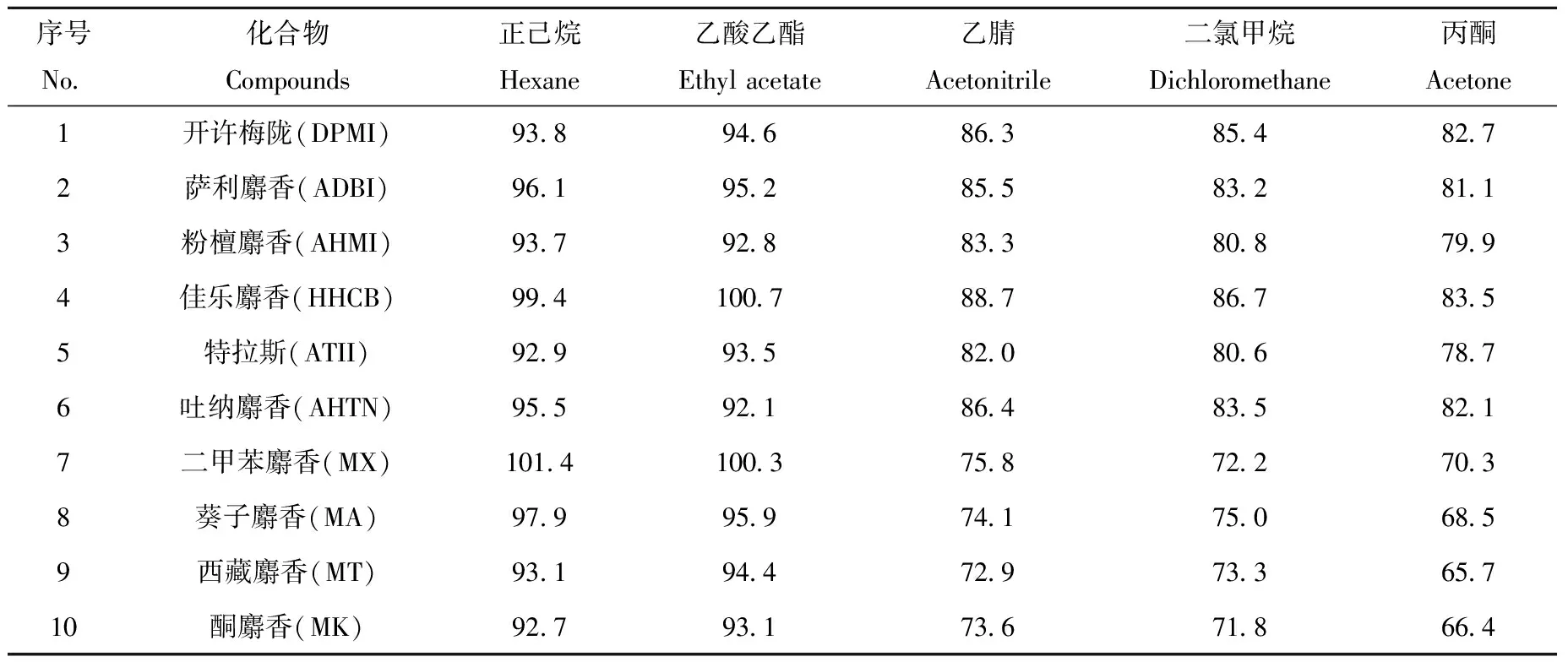
表2 使用不同提取溶剂时,膏霜类化妆品中10种合成麝香的回收率(n=6)Table 2 Recoveries of 10 synthetic musks in cream with different extraction solvents (n=6)
2.1.3提取方式的优化 综合考虑提取方式的方便性与快捷性,实验考察了振荡提取、涡旋提取和超声提取3种方式的提取效率,结果表明,超声提取方法的提取效率最好。进一步考察了不同提取温度和超声时间对提取效率的影响。由于膏霜类样品的粘度较大,因此以膏状化妆品为加标样品,加入1.0 mL饱和氯化钠水溶液和2.0 mL正己烷,涡旋混匀后,经超声提取,结果示于图1。在10~40 ℃区间,合成麝香的回收率随着超声温度的上升而不断提高,但温度超出40 ℃后,合成麝香的回收率反而呈现轻微下降,这与DPMI、ADBI等几种低沸点易挥发的麝香化合物有关,温度越高越易造成该类物质的损失。由图1b可见,回收率在前20 min内一直呈上升趋势,20 min后趋于平缓,且在20~40 min内合成麝香的回收率没有显著性差异。因此,在40 ℃下超声提取20 min,对化妆品样品中10种合成麝香的提取效率最高。(注:由于篇幅所限,图1只给出了所有合成麝香待测物的平均回收率趋势,若需10 种物质的详细曲线和数据,请联系作者)

图1 超声温度(a)和超声时间(b)对回收率的影响Fig.1 Effects of ultrasonic temperatures (a) and ultrasonic times (b) on recoveries
2.2 仪器条件的优化
2.2.1色谱柱的选择 通过参考相关文献,并考虑方法的普及性,本实验首先选用了气相色谱-质谱联用仪最常用的非极性毛细管色谱柱DB-5MS(或同类型填料柱)。化合物在DB-5MS色谱柱上基本按照其沸点由低到高的顺序出峰,因为HHCB、ATII、AHTN和MX的沸点差异不大,所以它们在该色谱柱上的保留时间很接近,通过优化程序升温条件仍然无法实现完全分离。实验选择了同样规格但不同填料的DB-1701、VF-WAXms和HP-FFAP三种色谱柱,希望利用化合物极性的差异,通过改变毛细管柱填料极性实现化合物的分离。结果表明,VF-WAXms和HP-FFAP的分离效果较好,各物质均能实现完全分离,但是由于HP-FFAP是非质谱柱,在高温下使用时会有较大的柱流失,易造成质谱污染。因此,本实验最终选择VF-WAXms色谱柱。在MRM扫描模式下,10种合成麝香和2种氘代内标在VF-WAXms柱上的色谱图示于图2。图中6、7号峰是d3-AHTN和AHTN的重叠峰,虽然二者存在共流出,无法完全分开,但在定性定量时可通过提取离子对进行选择性地分离,不影响测定结果。另外,图中的4号峰有2个基本分离的峰,这是HHCB的2个同分异构体,在定量计算时将这2个异构体峰作加和处理。
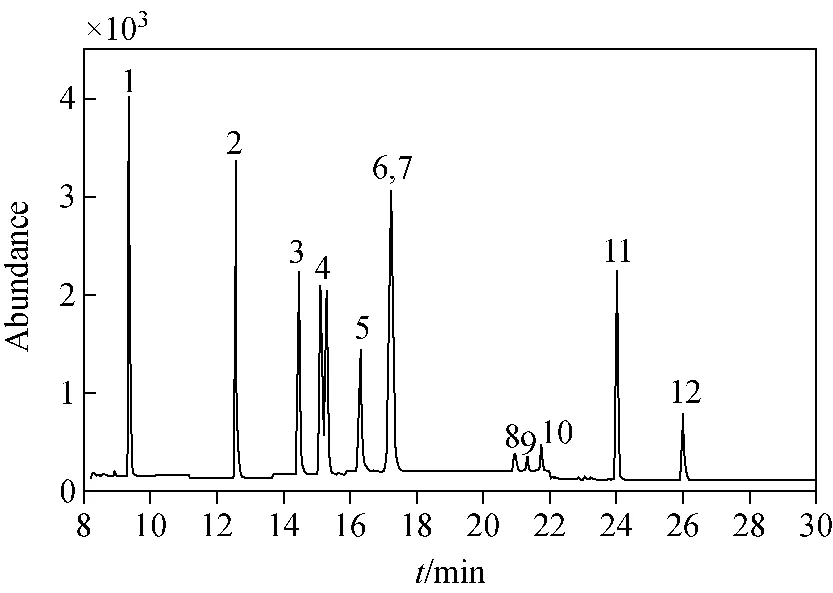
注:1.DPMI;2.ADBI;3.AHMI;4.HHCB;5.ATII;6,7.d3-AHTN,AHTN;8.d15-MX;9.MX;10.MA;11.MT;12.MK图2 10种合成麝香(5.0 μg/L)和2种氘代内标的MRM色谱图Fig.2 MRM chromatograms of 10 synthetic musks (5.0 μg/L) and 2 deuterium-labeled isotope internal standards
2.2.2三重四极杆串联质谱条件的建立 由于合成麝香的电负性较弱,因此选择EI质谱电离模式,以MRM 模式扫描。基于GC-MS/MS 中母离子和子离子的对应关系,通过设定6个时间段和多个扫描通道同时检测10种合成麝香和2种氘代内标。
首先,通过气相色谱和单极全扫描确定化合物的保留时间和一级碎片离子,选择响应值较好的一级碎片离子作为母离子。然后选择合适的碰撞电压,使合成麝香母离子与碰撞气体(N2) 进行碰撞诱导解离(CID),进一步裂解产生合成麝香碎片子离子,通过质量分析器扫描分析得到合成麝香的二级质谱图。分别选取丰度较强的主要碎片离子作为定量子离子,丰度次强的主要碎片离子作为辅助定性子离子。通过优化碰撞能量使目标合成麝香的基峰离子与特征碎片离子产生的离子对强度均达到最大,设定产生最强子离子响应的碰撞电压为最佳碰撞电压,具体参数列于表1。
2.3 检出限与标准曲线
按照优化后的实验条件考察方法的检出限。称取1.00 g 样品,提取定容至1.0 mL,进样量1 μL,以被测物质的信噪比S/N≥3时的进样浓度为检出限,以S/N≥10时的进样浓度为定量限。
采用内标标准曲线法定量测定。以目标化合物峰面积与内标物峰面积的比值为纵坐标,以标准溶液浓度与内标物浓度的比值为横坐标,绘制标准曲线。6种多环麝香(DPMI、ADBI、AHMI、HHCB、ATII和AHTN)以d3-AHTN为内标,4种硝基麝香(MX、MA、MT和MK)以d15-MX为内标进行校正。10种合成麝香在各自的浓度范围内线性关系良好,相关系数(R2)均大于0.99,各化合物的回归方程、线性相关系数、线性范围和定量限列于表3。
2.4 准确度和精密度实验
向不含合成麝香的空白膏霜化妆品样品中分别加入低、中、高3个质量浓度的10种合成麝香标准样品,按1.3.1节前处理方法提取并平行测定6次,计算10种合成麝香的加标回收率及方法精密度。结果表明,该方法的回收率为92.5%~102.0%,RSD为1.14%~4.50%,详细数据列于表4。
2.5 实际样品分析
利用本实验建立的方法检测市售的100种水、乳液和膏霜类化妆品。结果表明,在所检的100批次样品中,共有43批次检出合成麝香类物质,检出率较高。其中检出率最高的是HHCB,达36%,HHCB在一款保湿面膜中的检出含量高达499.9 mg/kg;其次是AHTN,检出率为27%,其含量范围为0.1~119.2 mg/kg;在6批次样品中检出DPMI,含量范围为3.2~225.8 mg/kg;在4批次样品检出ATII,含量范围为0.2~24.0 mg/kg。以上被高频率检出的均为多环麝香,目前国内还没有出台任何法规约束其使用范围或使用量。ADBI、AHMI、MK和MA各有1批次样品检出,含量分别为5.6、0.8、5.0和92.1 mg/kg。值得注意的是,MA虽已被全面禁用,但仍然存在不法商家违规添加的现象。已禁用的MX和MT在所有样品中均未检出。
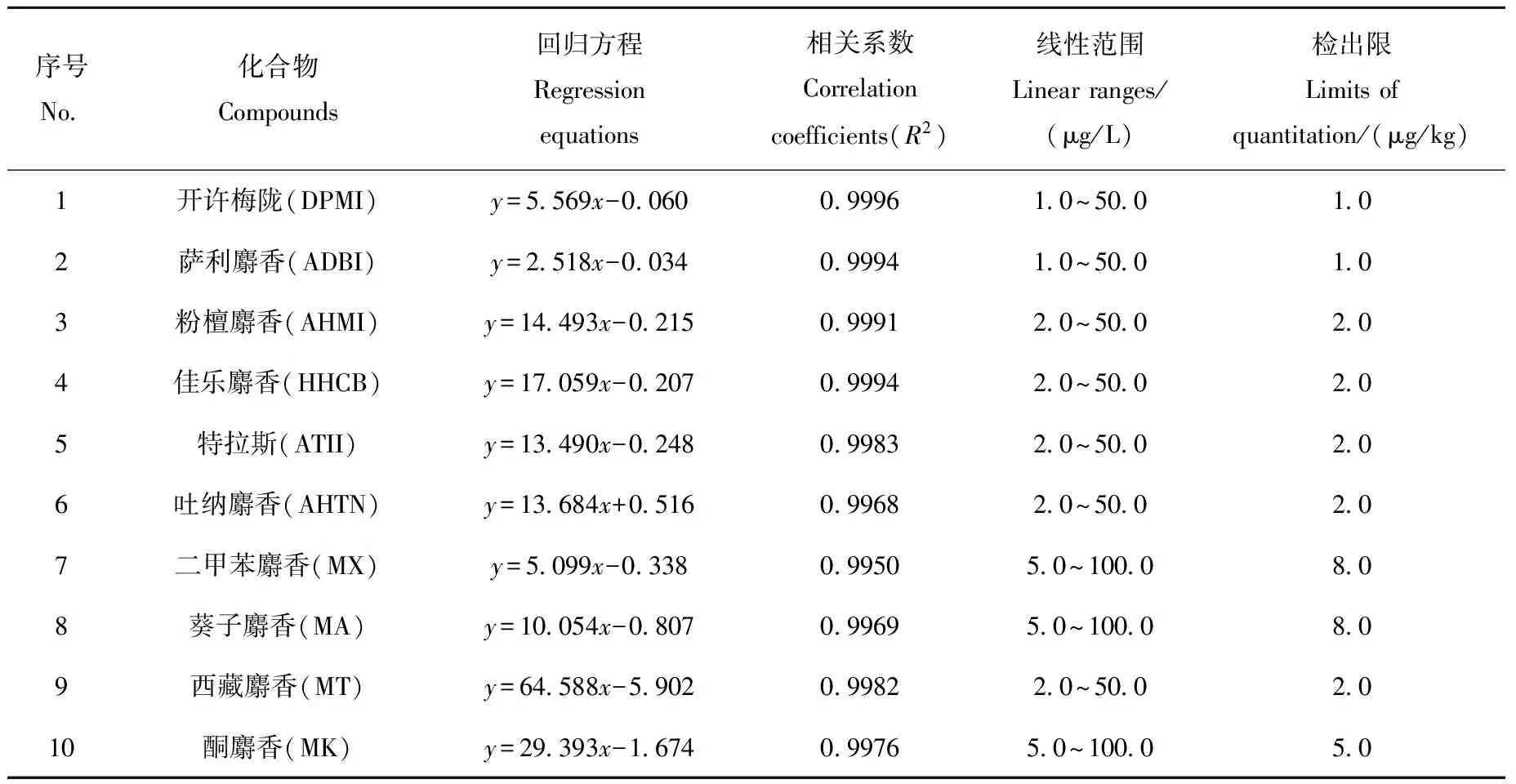
表3 10种合成麝香的回归方程、相关系数、线性范围和定量限Table 3 Regression equations, correlation coefficients, linear ranges and limits of quantitation of 10 synthetic musks

表4 10种合成麝香的加标回收率和相对标准偏差(n=6)Table 4 Spiked recoveries and RSDs of 10 synthetic musks (n=6)
3 结论
目前,化妆品中合成麝香的分析检测方法尚处于起步阶段。本研究建立了GC-MS/MS法快速检测10种合成麝香,在1.0~100.0 μg/L质量浓度范围内,10种合成麝香的仪器响应值与浓度呈显著的线性相关,相关系数均大于0.99;方法定量限达1.0~8.0 μg/kg。10种合成麝香的加标回收率在92.5%~102.0%之间,相对标准偏差(RSD)均小于5%。该方法简便、准确、快速、灵敏度高、重现性好,可同时检测化妆品中10种合成麝香,适合标准化推广,对香精香料、日化行业的产品质量控制具有重要意义。
