先天性皮肤发育不全基因检测一例
2018-07-07樊晓艳李慧汪佳杨丽吴卉
樊晓艳 李慧 汪佳 杨丽 吴卉
225300江苏,泰州市人民医院新生儿科(樊晓艳、李慧、杨丽),生殖医学科(汪佳),皮肤科(吴卉)
先天性皮肤发育不全是出生时部分表皮、真皮和(或)皮下组织缺如[1],其病因尚不完全明确,临床无特效方法治疗。现将我们诊治的1例报道如下。
一、病历资料
患儿女,出生2 h,生后即发现皮肤缺损并水疱2 h,于2016年11月由外院转入泰州市人民医院新生儿科。患儿系第3胎第2产,孕39周择期剖宫产,出生体重3 450 g,Apgar评分9~10分/1~5 min,羊水清,脐带及胎盘无异常。其母妊娠3次,人工流产1次,2015年9月孕34周余不明原因胎死宫内行引产术,死胎娩出时皮肤有破损,尸检除皮肤破损外未见异常。本次妊娠系单胎妊娠,孕期无感染史,无有毒物或放射线接触史,定期产检无异常。父母非近亲婚配,家族中无类似患者。体检:一般情况好,各系统检查未见异常。皮肤科检查:双下肢及左手腕皮肤成片不对称、不规则缺损(图1),面积约占体表面积10%,表面可见一层薄膜,基底潮红,无出血及渗液,血管走行清晰,缺损与正常皮肤界限清楚,周围皮肤正常,右手手背可见一薄壁张力性水疱,3 cm×4 cm,疱液清亮,尼氏征阴性。
实验室检查:血常规、C反应蛋白、血气分析正常。Torch抗体IgM、抗肝炎病毒等、梅毒及抗HIV抗体检查均无异常。疱液培养、血液培养72 h无异常。在家长签署知情同意书后对患儿及其父母行染色体、基因检测。基因检测方法用乙二胺四乙酸(EDTA)抗凝管抽取患儿及父母外周静脉血各2 ml。采用QIAamp DNA提取试剂盒(QIAGEN公司)抽提基因组DNA,并测量其吸光度值及浓度。提取的DNA用DNA酶片段化后用磁珠法进行纯化,随后进行PCR扩增并连接上接头序列,经定制的Panel探针(illumina Inc,USA)两次捕获及纯化,再经PCR扩增和纯化后获得的最终文库在NextSeq500测序仪(illumina Inc,USA)上对Panel相关基因的外显子区进行测序。对可疑候选突变的位点设计PCR引物进行扩增及进行Sanger测序验证,并对患儿父母相应位点进行检测。出院后染色体核型检查结果:46,XY;基因检测结果报告(命名方式遵从HGVS国际规范)提示COL7A1(NM⁃000094)exon4:c.481c>T;P.(Gln161*)杂合,致病突变;COL7A1(NM⁃000094)exon14:c.1837C > T;P.(Arg613*)杂合,致病突变。对其父母亲COL7A1(NM⁃000094)基因的C.481位点和C.1837位点进行检测,父亲检测结果示COL7A1(NM⁃000094)exon4:c.481c > T;P.(Gln161*)杂合;母亲检测结果示COL7A1(NM⁃000094)exon14:c.1837C > T;P.(Arg613*)杂合(图2)。
治疗:因家长拒绝行皮肤活检,故临床诊断先天性皮肤发育不全。入院后予保护性隔离,生理氯化钠溶液局部冲洗,并外用表皮生长因子;头孢他啶防治感染,住院治疗6 d,患儿吃奶好,呼吸平稳,生命体征平稳,皮损病变无明显变化,自动出院。住院期间右手背疱疹破溃,出院时表皮缺失部位基底部潮红,创面干燥无渗出。出院后家长自予表皮生长因子局部应用,出院1个月电话随访,皮肤缺损部位渐干燥,色泽发灰,后失访。

图1 患儿临床皮损表现
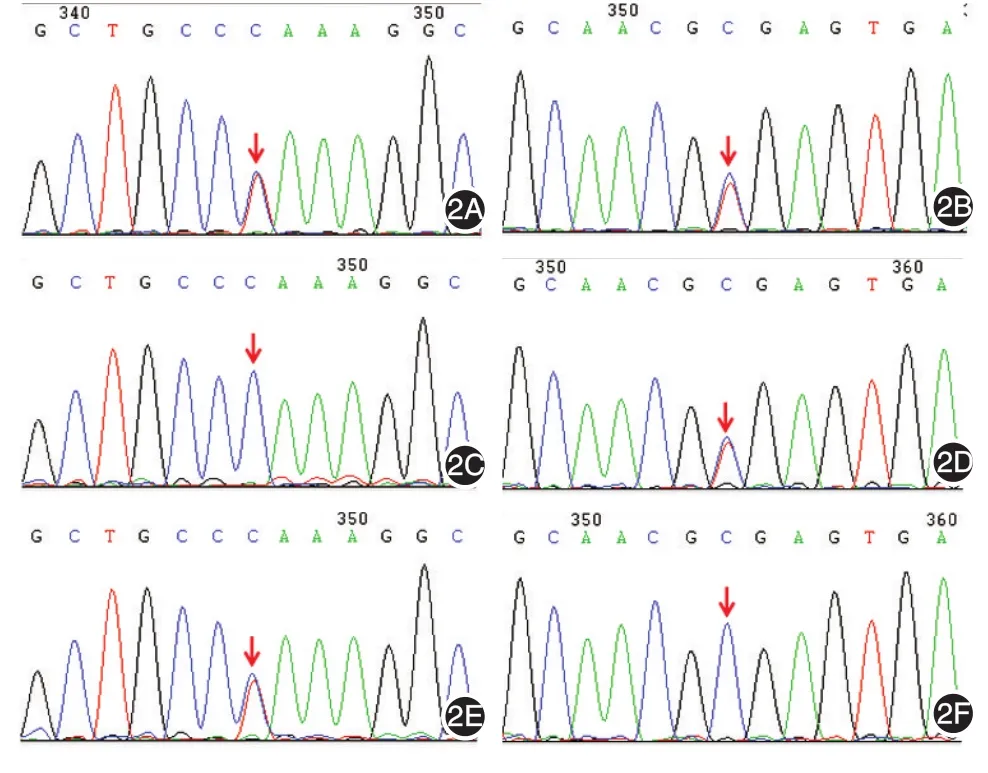
图2 患儿及父母COL7A1基因c.481c和c.1837c位点检测2A:患儿COL7A1基因c.481c>T杂合突变;2B:患儿父亲COL7A1基因c.481c位点未见异常;2C:患儿母亲COL7A1基因c.481c>T杂合突变;2D:患儿COL7A1基因c.1837c>T杂合突变;2E:患儿父亲COL7A1基因c.1837c>T杂合突变;2F:患儿母亲COL7A1基因c.1837c位点未见异常
二、讨论
先天性皮肤发育不全病因不明,目前倾向于胎儿宫内缺损部位血管发育异常致局部血流中断引起的病变,不能用单一理论完全解释所有先天皮肤缺损疾病。主要认为与染色体异常、基因病(常染色体显性或隐性遗传、X连锁)、子宫腔狭小、宫腔内压力高、胎儿皮肤与羊膜粘连及双胎其一死亡引起血栓栓塞[2]等因素相关。本例患儿出生时皮肤缺损,其病变限于四肢,伴大疱,根据弗里登ACC分类,该患儿属第Ⅵ类,即皮损位于四肢,伴有大疱性表皮松解症,水疱多位于四肢,与隐性遗传病例特征相符。临床诊断为常染色体隐性遗传先天性皮肤发育不全伴营养不良性大疱性表皮松解症。有报道认为,此病由一组常染色体隐性基因控制,此基因为整合系基因,可以影响基底膜和半桥粒的完整性,并控制伤口愈合过程中纤维化的正常进程。本例患儿单胎妊娠,定期产检,孕期无感染史,无有毒物质接触史,病原学检查未见异常,可排除感染、双胎妊娠压迫、染色体异常等因素。基因检测显示,患儿存在COL7A1(NM⁃000094)基因的复合杂合病变,其父母亲分别存在COL7A1(NM⁃000094)基因的C.481位点和C.1837位点的杂合变异。COL7A1基因编码Ⅶ型胶原蛋白,位于3号染色体3p21.1,全长31.312 kb,其杂合致病突变可引起大疱性表皮松解症[3⁃4],COL7A1(NM⁃000094)exon4:c.481c>T;P.(Gln161*)为无义突变,预计所编码的蛋白质第161位氨基酸Gln变为终止密码子,并导致翻译提前终止,该突变尚未见文献报道,ESP6500siv2_ALL、千人基因组(1000g2015aug_ALL)和dbSNP147数据库均未收录。COL7A1(NM⁃000094)exon14:c.1837C>T;P.(Arg613*),预计所编码的蛋白质第613位氨基酸Arg变为终止密码子,并导致翻译提前终止。有报道在大疱性表皮松解症患者中检测到该变异,ESP6500siv2_ALL、千人基因组(1000g2015aug_ALL)数据库均未收录,dbSNP147(rs759634066)有收录[5]。本例患儿孕母有死胎史1例,死胎剖宫产时伴皮肤缺损。患儿出生时皮肤缺损,基因检测有2个可疑的致病突变,其父母分别检测到2个位点之一的异常,但均无临床病变表现,推测COL7A1(NM⁃000094)基因C.481位点和C.1837位点的杂合异常组成复合杂合子,为患儿致病的基因突变,此突变可能干扰Ⅶ型胶原的三螺旋结构域的形成,影响了其结构的完整性和稳定性,致皮肤基底膜致密板下带锚纤维(其主要成分为Ⅶ型胶原)不能维持正常的功能,致患儿发病。该病目前尚无特效治疗,主要原则是保护创面,预防和治疗感染,促进创面愈合。对检测到的两个突变位点我们将继续进行健康人群的验证,从而明确此突变的意义。由于本病无特效治疗,以患儿的致病基因为先证进一步研究有助于宫内DNA分析,实现产前诊断和优生优育。
[1]陈明,潘红梅.皮肤再生不良合并大疱性表皮松解症及先天性截趾一例[J].中华皮肤科杂志,2006,39(4):230.doi:10.3760/j.issn:0412⁃4030.2006.04.026.
[2]陈钦标,柳国胜.双卵双胎新生儿先天性皮肤缺损1例[J].临床荟萃,2015,(2):223⁃224.doi:10.3969/j.issn.1004⁃583X.2015.02.024.
[3]刘宁,郭宏湘,孔祥东,等.营养不良性大疱性表皮松解症COL7A1基因诊断及产前诊断[J].中华医学杂志,2015,95(4):277⁃282.doi:10.3760/cma.j.issn.0376⁃2491.2015.04.009.
[4]于灵,冯素英.先天性大疱性表皮松解症诊断及治疗进展[J].中华皮肤科杂志,2016,49(7):516⁃519.doi:10.3760/cma.j.issn.0412⁃4030.2016.07.019.
[5]Varki R,Sadowski S,Uitto J,et al.Epidermolysis bullosa.II.Type VII collagen mutations and phenotype⁃genotype correlations in the dystrophic subtypes[J].J Med Genet,2007,44(3):181⁃192.doi:10.1136/jmg.2006.045302.
