乌及丸的质量控制方法研究
2018-07-05王东风陈明明许志伟
赵 昕,张 鹏,黄 娜,王东风,陈明明*,许志伟
0 引言
乌及丸是解放军第205医院配制的非标准制剂,由白芍、白及、高良姜、甘草、延胡索、海螵蛸、香附等药味组成,有温中散寒、行气止痛、制酸止血的功效,用于慢性胃炎、胃及十二指肠溃疡、胃酸过多等症,在临床上疗效满意。为保证临床用药有效、安全,对本研究乌及丸进行了质量控制研究,制定了乌及丸的检验方法,制订后的定性定量方法可控制乌及丸的质量。
1 仪器与试药
高效液相色谱仪: LC-20AD高效液相色谱仪(日本岛津);KQ3200E型昆山超声波处理器。电子分析天平(120D型,北京岛津公司);对照药材(中检院,延胡索: 110726-201414;高良姜:121263-201304);对照品(中检院,甘草次酸:110723-201413;芍药苷:供含量测定用,110736-201438);乌及丸(由解放军第205医院提供,批号:20141120、20141204、20141218);甲醇(色谱纯,Fisher公司),乙腈(色谱纯,Sigma公司),磷酸(分析纯,天津市瑞金特化学品有限公司),重蒸水(自制)。
2 方法与结果
2.1 显微鉴别
2.1.1 海螵蛸 显微镜下可见透明碎片呈不规则形状且其上有网状或点状纹理。见图1。

图1 不规则透明碎片(海螵蛸)
2.1.2 高良姜 置显微镜下淀粉粒棒槌形、肾形或菱角形,有的中部突出作分枝状,长约至50 μm,稀有更长的,脐点点状、短缝状或不明显。见图2。

图2 淀粉粒(高良姜)
2.1.3 白及 置显微镜下观察:表皮细胞观垂周壁波状弯曲,略增厚。见图3。
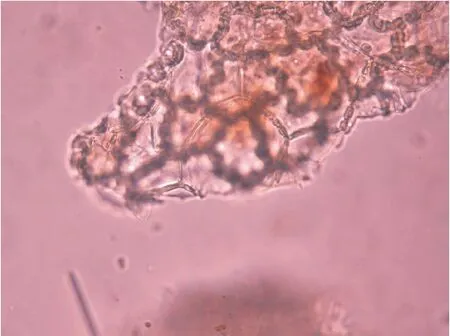
图3 表皮细胞(白及)
2.2 薄层色谱鉴别
2.2.1 延胡索[1]取乌及丸1丸,剪碎,加硅藻土适量研磨成粉末,加浓氨试液适量使润湿,加二氯甲烷50 ml加热回流1 h,放至室温,过滤,滤液蒸干,加二氯甲烷1 ml使溶解,作为供试品溶液;另取延胡索对照药材粉末约2 g,按供试品溶液制备方法操作,制得对照药材溶液。按乌及丸制备工艺,制备乌及丸缺延胡索阴性样品,取1丸,按供试品溶液制备方法操作,制得乌及丸缺延胡索阴性样品溶液。照薄层色谱法[2],取上述3种溶液各5 μl,分别点于硅胶G薄层板上,置层析缸中,展开剂为乙酸乙酯-甲酸-乙酸-水(10∶1∶1.5∶2),展开,取出,晾干,在紫外光灯(365 nm)下检视,在乌及丸供试品色谱与延胡索对照药材色谱相应的位置上,显相同颜色的荧光斑点,阴性对照无干扰(图4)。此外,3批样品荧光斑点重现性较好(图5)。
2.2.2 高良姜[3]取乌及丸2丸,剪碎,加硅藻土8 g研粉,加二氯甲烷50 ml超声30 min,过滤,滤液蒸干,加二氯甲烷1 ml 使溶解,作为供试品溶液;另取高良姜对照药材研粉末5 g,按供试品溶液制备方法操作,制得对照药材溶液;按乌及丸制备工艺,制备乌及丸缺高良姜阴性样品,取2丸,按供试品溶液制备方法操作,制得乌及丸缺高良姜阴性样品溶液。照薄层色谱法[2]取上述3种溶液各10 μl,分别点于硅胶G薄层板上,置层析缸中,展开剂为石油醚(60~90 ℃)-乙酸乙酯-甲酸(9∶1∶0.1),展开,取出,晾干,用5%香草醛硫酸溶液显色,放于干燥箱内105 ℃加热至斑点清晰。乌及丸供试品与高良姜对照药材色谱相同位置上显相同颜色的斑点,阴性对照无此斑点(图6)。此外,3批样品斑点重现性较好(图7)。

图4 延胡索薄层色谱鉴别(专属性)
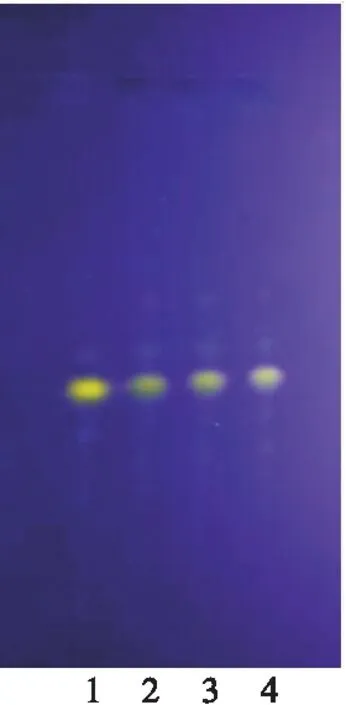
图5 延胡索薄层色谱鉴别(重复性)

图6 高良姜薄层色谱鉴别(专属性)
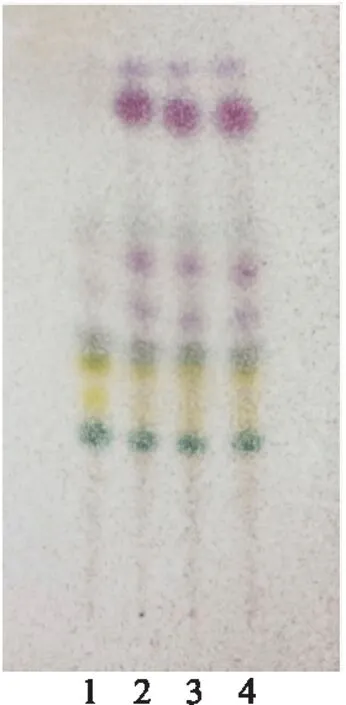
图7 高良姜薄层色谱鉴别(重复性)
2.2.3 甘草[1,4]取乌及丸1丸,剪碎,加4 g硅藻土研磨成粉末,加盐酸10 ml及二氯甲烷50 ml加热回流1 h,放至室温,过滤,滤液蒸干,加二氯甲烷1 ml使溶解,作为供试品溶液;精密称取甘草次酸对照品20 mg于10 ml容量瓶中,加入甲醇至刻度,作为甘草次酸对照品溶液;按乌及丸制备工艺,制备乌及丸缺甘草阴性样品,取1丸,按供试品溶液制备方法操作,制得乌及丸缺甘草阴性样品溶液。参照薄层色谱法[2],制得乌及丸缺甘草的阴性样品溶液。参照薄层色谱法[2],取上述3种溶液各10 μl,分别点于硅胶GF254薄层板上,置层析缸中,展开剂为环己烷-乙酸乙酯-冰醋酸(3∶1∶0.2),展开,取出,晾干,在紫外光灯(254 nm)下检视,乌及丸供试品色谱与甘草次酸对照品色谱相应的位置上,显相同深蓝色的斑点,阴性对照无此斑点(图8)。此外,3批样品此斑点重现性较好(图9)。
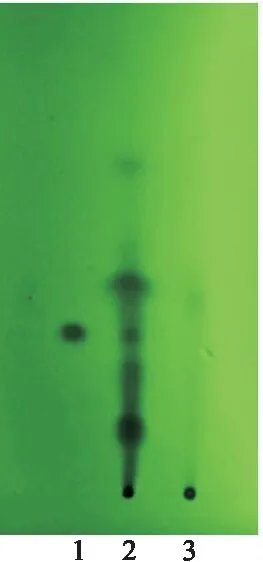
图8 甘草薄层色谱鉴别 (专属性)
2.3 乌及丸中芍药苷含量测定[1,5-6]
2.3.1 溶液的制备 ①对照品溶液的制备:精密称取芍药苷对照品(已置五氧化二磷减压干燥器中12 h)20 mg于500 ml容量瓶中,加入适量稀乙醇使溶解并稀释至刻度,作为对照品溶液。②乌及丸样品溶液的制备:取乌及丸1.5 g,剪碎,精密称定,加硅藻土适量,研磨成粉末,放于已精密加入50 ml稀乙醇的三角瓶中,密塞后称重,超声30 min,放至室温,称重并补充稀乙醇至超声前重量,过滤,取续滤液,作为乌及丸样品溶液。③阴性样品溶液的制备:取除白芍外的其他药材,按乌及丸处方比例及制剂工艺制成丸剂,按照“2.3.2”项方法制成阴性样品溶液。
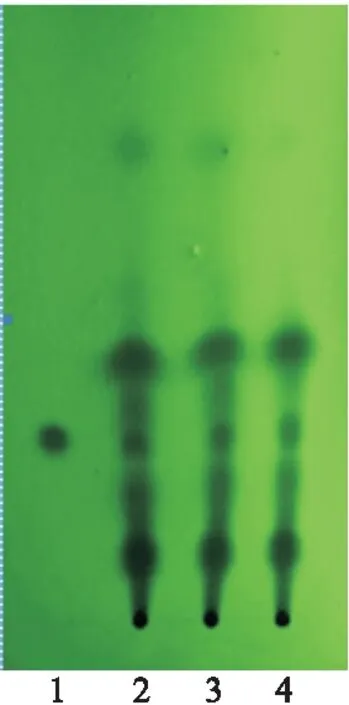
图9 甘草薄层色谱鉴别(重复性)
2.3.2 色谱条件与专属性 VP-ODS (5 μm,4.6 mm×150 mm) 色谱柱;乙腈-0.1%磷酸(15∶85)为流动相;柱温:30 ℃;流速:1 ml/min;在230 nm波长下检测;各种溶液分别进样10 μl;理论板数按芍药苷峰计算不低于2 000。分别取上述3种溶液进样,结果表明,样品中待测成分分离良好,阴性对照无干扰,见图10。
2.3.3 芍药苷对照品标准曲线 在100 ml容量瓶中精密称取芍药苷对照品(已置五氧化二磷减压干燥器中12 h)25 mg,加入稀乙醇使溶解并稀释至刻度,配制成250 μg/ml芍药苷对照品溶液,分别精密取此芍药苷对照品溶液0.5、1、2、4、8、16 ml于50 ml容量瓶中,加入稀乙醇使溶解并稀释至刻度,配制成2.5、5、10、20、40、80 μg/ml的溶液,注入液相色谱仪各10 μl,按“2.3.2”项方法测定各自峰面积,以对照品浓度(μg/ml)为横坐标,峰面积为纵坐标,计算得回归方程Y=5 456.1X-4 956.7,r=0.999 4。结果可见测得峰面积与芍药苷对照品在2.5~80 μg/ml范围内线性关系良好。
2.3.4 精密度 精密吸取芍药苷对照品溶液10 μl,按“2.3.2”项方法连续进样6次,结果峰面积值的RSD为0.45% (n=6),符合要求。
2.3.5 稳定性 取同一批乌及丸样品约1.5 g,剪碎,精密称定,加硅藻土适量,研磨成粉末,按照“2.3.1”项操作制备供试品溶液,取续滤液按“2.3.2”项分别在0、2、4、6、8 h进样,测得芍药苷对照品峰面积值的RSD为1.81% (n=5),表明其在8 h内稳定。
2.3.6 重复性 取同一批乌及丸约1.5 g(共6份),精密称定,加硅藻土适量,研磨成粉末,按照“2.3.1”项操作制备供试品溶液,取续滤液按“2.3.2”项方法测定。结果样品中芍药苷平均含量为0.77 mg/g,RSD为1.68% (n=6),符合要求。
2.3.7 加样回收率 分别精密称定同一批共6份乌及丸(每份约0.7 g),分别剪碎后,加入芍药苷对照品溶液(560 μg/ml)各1 ml,称定重量后加硅藻土适量,研磨成粉末,放于已精密加入50 ml稀乙醇的三角瓶中,密塞后称重,超声30 min,放至室温,称重并补足重量,取其续滤液,按“2.3.2”项方法测定样品中芍药苷含量,结果符合要求。见表1。
2.3.8 样品测定 取3个不同批号(批号:20141120、20141204、20141218)的乌及丸样品,按“2.3.1”项操作,按“2.3.2”测定其中芍药苷的含量。结果见表2。
3 讨论
3.1 《中国药典》及文献中延胡索的薄层色谱鉴别多采用较大比例的甲醇作为提取溶剂,因其对实验操作人员危害较大,故在本实验中经多次试验将其替换为毒性较小的二氯甲烷作为提取溶剂,达到同样提取效果的同时降低了对实验操作人员健康的损害。
3.2 乌及丸中芍药苷含量测定中,制备样品溶液时,为了保证回收率,本实验分别对提取方式、提取溶剂、溶剂用量、提取时间及提取次数进行考察。其中考察提取方式时,分别采用浸泡、超声提取与加热回流的方式,结果表明,采用超声和加热提取方法时,二者所测得的芍药苷含量较高且含量相当,故将提取方法定为操作更为简便、重现性更好的超声提取;考察提取溶剂时,分别使用了30%乙醇、稀乙醇、甲醇、30%甲醇等溶剂,结果表明,提取溶剂为稀乙醇及甲醇时所测得的芍药苷含量近似且高于其他溶剂,故采用稀乙醇为提取溶剂,结果满意。

图10 芍药苷高效液相色谱图
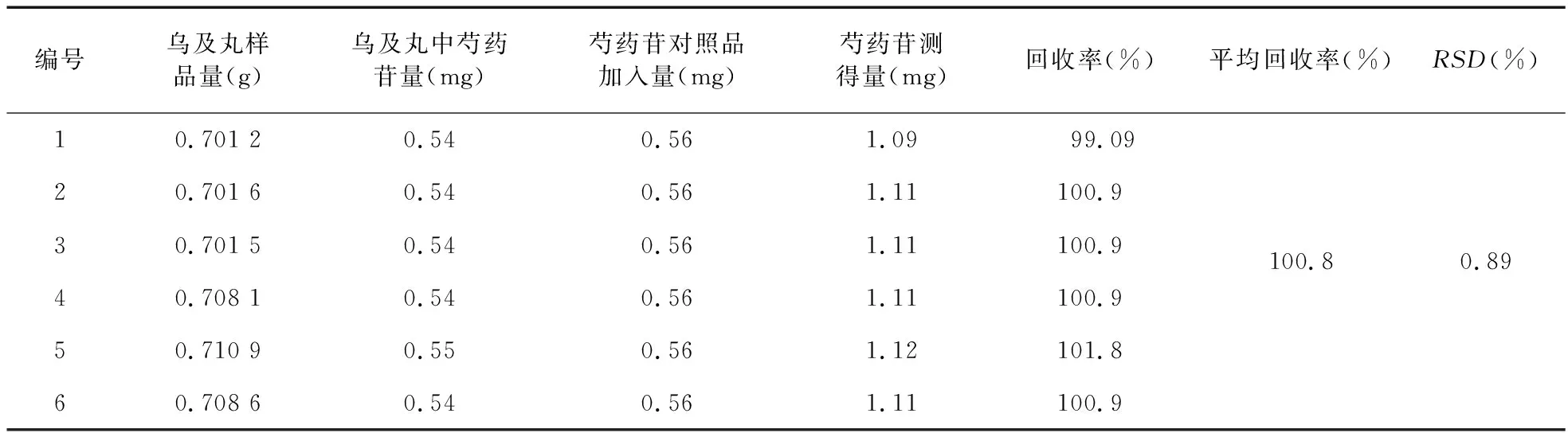
编号乌及丸样品量(g)乌及丸中芍药苷量(mg)芍药苷对照品加入量(mg)芍药苷测得量(mg)回收率(%)平均回收率(%)RSD(%)10.701 20.540.561.0999.0920.701 60.540.561.11100.930.701 50.540.561.11100.9100.80.8940.708 10.540.561.11100.950.710 90.550.561.12101.860.708 60.540.561.11100.9

表2 乌及丸含量测定结果
参考文献:
[1] 国家药典委员会.中华人民共和国药典(一部)[S].北京:中国医药科技出版社,2015:86-87,139-140,901-902.
[2] 国家药典委员会.中华人民共和国药典.(四部)[S].北京:中国医药科技出版社,2015:57-58.
[3] 米仁沙·牙库甫,热娜·卡斯木,努尔阿尼也·热合曼,等.小艾飞蜜膏质量标准研究[J].新疆医科大学学报,2014,37(4):436-439.
[4] 于妮娜,张玲.薄层色谱法鉴别归芪养血益气口服液中甘草的研究[J].现代中医药,2016,36(4):80-82.
[5] 彭俊付,赵宝明,张书信,等.RP-HPLC测定广痛消中芍药苷含量的方法[J].中国中药杂志,2011,36(23):3268-3269.
[6] 曾棋平,杨丽娜,王艺红,等.气血双补口服液的稳定性研究[J].药学服务与研究,2017,17(5):384-386.
