7-氯-4-羟基二氢化茚的合成研究
2018-07-05赵长阔王先恒
赵长阔,王先恒,袁 智,曹 颖,徐 浪
(遵义医学院 药学院,贵州 遵义 563099)
茚是医药及化工领域中重要的一类化学结构,其衍生物主要包括茚、二氢化茚、茚酮及茚醇等广泛存在于天然产物、药物、农药等生物活性分子[1-5]。目前已有多个茚类药物上市,抗抑郁药物茚达曲林(indatralline)、用于成人慢性阻塞性肺疾病患者的维持治疗的支气管扩张剂茚达特罗为二氢化茚胺化合物;治疗老年痴呆症的多奈哌齐(Donepezil)和治疗HIV感染和艾滋病的蛋白酶抑制剂茚地那韦(Indinavir)为茚酮类药物[2];茚并吡唑 (GN39484)属于微管蛋白抑制剂,具有较好的抗恶性肿瘤活性。
最近的研究发现,7-氯-4-羟基二氢化茚是合成许多活性药物分子的重要结构片段[6-8]。例如,7-氯-4-羟基二氢化茚的化合物Ia具有胆固醇酯转运蛋白(CETP)抑制活性,抑制CETP蛋白可抑制胆固醇酯从高密度脂蛋白(HDL)转移到低密度脂蛋白(LDL)和极低密度脂蛋白(VLDL),从而导致HDL的增高以抑制动脉粥样硬化[6]。辉瑞制药(Pfizer)报道的苯并哌嗪类的化合物具有γ-分泌酶调节活性,能够可穿透血脑屏障,有潜力用于阿兹海默症的治疗[8]。

文献对于7-氯-4-羟基二氢化茚的合成报道不多:Buck J S报道了使用4-羟基二氢化茚a在冰醋酸溶液中与二氯亚砜在55℃反应1 h得到5-及7-位单氯代混合产物,由于氯代反应对于苯环邻、对位置选择性不佳,7-氯-4-羟基二氢化茚1 收率仅为 47%[9](Scheme 1)。

Scheme 1 4-氯-7-羟基二氢化茚1的合成路线[9]
Am Ende CW在专利文献WO2012131539A1中报道了将1-氧代-4-羟基二氢化茚b用氰基硼氢化钠还原后用三甲基氯硅烷处理后,得到4-羟基二氢化茚c,再用氯代琥珀酰亚胺氯化后得到化合物1,但没有报道产物收率[10]。

Scheme 2 7-氯-4-羟基二氢化茚1的合成路线[10]
由于上述两种合成方法中均是以4-羟基二氢化茚衍生物作为原料,原料昂贵也不易制备获得;因此本研究尝试了一种新的便宜的合成方法:以对氯苯酚为起始原料,依次通过酚羟基酯化、Fries酰基重排后亲电取代得到茚环、酮羰基还原三步骤反应得到7-氯-4-羟基二氢化茚 (化合物1),总收率高达46.2%。
1 材料与仪器
1.1 材料
对氯苯酚 (分析纯,Adrich)、β-氯丙酰氯、三乙基硅烷、浓盐酸(35%)(分析纯,国药集团化学试剂有限公司);无水氯化铝、三氟乙酸、氢氧化钾、氢氧化钠、无水硫酸钠、无水硫酸镁 (化学纯,国药集团);二氯甲烷、氯仿、甲醇、乙醇、乙酸乙酯、石油醚、己烷(化学纯,成都市科龙化工试剂厂);硅胶薄层板(青岛海洋化工厂)、柱层析用硅胶(200~300 目);自制纯化水。
1.2 仪器
Varian 400 MHz核磁共振波谱仪 (TMS作内标,安捷伦科技有限公司);LC-10ATVP型高效液相色谱仪(日本岛津公司);电热恒温鼓风干燥箱(上海精宏实验设备有限公司,DMG-914M型);暗箱三用紫外分析仪 (上海司乐仪器有限公司,81-2型)。
2 合成方法与讨论
2.1 化合物1的合成路线
以对氯苯酚为起始原料,依次通过酚羟基酯化、Fries酰基重排后亲电取代得到茚环、酮羰基还原三步骤反应得到7-氯-4-羟基二氢化茚 (化合物1),总收率高达46.2%。
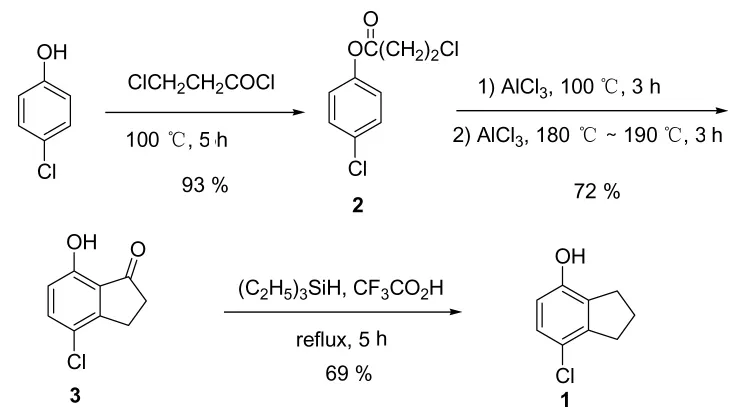
Scheme 3 化合物1的合成路线
2.1.1 对氯苯基 β-氯丙酸酯2的制备
在40℃~50℃下, 将 β-氯丙酰氯 (63 g,0.50 mol) 在搅拌下滴加至对氯苯酚 (64 g,0.50 mol)中;滴加完毕后,将反应混合物在 40 ℃~50℃下搅拌2 h,然后加热至100℃搅拌反应3 h。蒸馏所得反应油状混合物,得到化合物2(101.8 g,93% 产率), 为无色油状物。1H NMR(300 MHz, CDCl3) δ 7.41 (d, J=8Hz, 2H),7.12 (d, J=8Hz, 2H), 3.92 (t, 2H), 3.09 (t,2H); GC-MS 218[M]+.
2.1.2 7-氯-2,3-二氢-4-羟基茚-1-酮 3 的制备
将化合物 2 (138 g,0.63 mol)与无水氯化铝(250 g,1.87 mol)的混合物在 100 ℃下搅拌 3 h,然后升温至180℃~190℃继续搅拌反应3 h。将所得混合物倒入冰水中,用乙酸乙酯萃取三次(200 mL×3)。合并有机层,并用水洗涤直到pH为6~7,用无水MgSO4干燥。有机层浓缩后,所得残留物通过快速柱色谱纯化后得到化合物3,为黄色固体 (83 g,72%产率)。1H NMR (300 MHz,CDCl3) δ =9.00 (s, 1H), 7.47 (d,J=8 Hz,1H), 6.79 (d, J=8 Hz, 1H), 3.14 (t, 2H),2.80 (t, 2H); LC-MS 183[M+H]+.
2.1.3 7-氯-4-羟基二氢化茚 1 的制备
在氮气保护下,向化合物 3 (83 g,0.45 mol)的三氟乙酸 (300 mL)溶液中加入三乙基硅烷(160 g,1.38 mol),然后将反应溶液在加热回流温度下搅拌4 h。反应液减压除去溶剂后,将所得残留物倒入 aq.NaOH (2 mol/L,1 L)中,用己烷萃取3次 (300 mL×3)。将水层用浓盐酸酸化至pH=1,用乙酸乙酯萃取3次 (300 mL×3)。 合并有机层,用水洗涤并用无水Na2SO4干燥。有机层减压浓缩蒸除溶剂后,所得残余物用石油醚 (b.p.90℃~120℃)中结晶,得到化合物1,为白色固体 (53 g,69%产率):1H NMR (300 MHz, CDCl3)δ=7.03 (d, J=8Hz,1H), 6.59(d, J=8Hz, 1H),4.64 (s, 1H), 3.00~2.89(m, 4H), 2.19~2.09 (m,2H);HPLC 94.1%(Purity);GC-MS 168[M]+.
2.2 讨论
2.2.1 酯化反应
对于第一步的4-氯苯酚与β-氯丙酰氯的反应,虽然β-氯丙酰氯分子中存在两个氯原子,但是由于酰氯的活泼性,因此更加容易发生酯化反应,而不容易发生醚化反应。在工艺优化时,我们发现,反应温度对于反应收率有很大影响。在50℃反应24 h收率可达74%,而当温度升至100℃,反应5 h,收率高达93%。

Scheme 4 化合物1的酰氯合成法[9]

表1 温度对化合物2的影响Table 1 Temperature effect on compound 2
2.2.2 还原反应
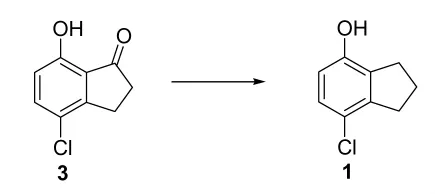
还原反应是整个反应路线当中最重要的一步反应,我们对于还原试剂的选择,溶剂的影响作了充分的研究。结果发现,7-氯-2,3-二氢-4-羟基茚-1-酮3在(CH3)3SiH还原条件下,以三氟乙酸为溶剂,得到目标化合物1。

表2 还原反应的工艺优化Table 2 Process optimization of reduction reaction
3 结论
本文发展了一条以对氯苯酚为起始原料,依次通过酚羟基酯化、酰基重排后亲电取代得到茚环、酮羰基还原三步骤反应得到7-氯-4-羟基二氢化茚1的新工艺,总收率高达46.2%。该方法为二氢化茚类衍生物的合成提供了一条新的选择。
[1] 段义杰,刘建利,王翠玲.茚酮类化合物的研究进展[J].有机化学,2010,30(7):988-996.
[2] 何桥,殷中琼,陈华保,等.茚及其衍生物的催化不对称合成[J].化学进展, 2016, 28(6): 801-813.
[3] 张圣淼,孙昊鹏,贾剑敏,等.具有抗肿瘤活性的茚并异喹啉类化合物研究进展[J].中国药科大学学报,2013,44(6): 589-595.
[4] 柴凤兰,徐海云.茚酮化合物合成方法研究进展[J].化工进展, 2014,33(11):3045-3052.
[5] 丛蔚,吴晓明,徐进宜.茚并异喹啉酮类拓扑异构酶1抑制剂的研究进展[J].药学与临床研究,2014,22(03):239-245.
[6] Harikrishnan L S,Kamau M G,Herpin T F,et al.2-Arylbenzoxazoles as novel cholesteryl ester transfer protein inhibitors:Optimization via array synthesis[J].Bioorg Med Chem Lett, 2008, 18: 2640–2644.
[7]Sugimoto T,Yamamoto K,Saitama K,et al.10A-Azalide compound crosslinked at position-10a and position-12:WO,2009019868A1[P].2009-02-12.
[8] Am Ende C W,Fish B A,Green M E,et al.Novel bicyclic pyridinones :WO,2012131539A1[P].2012-10-04
[9] Buck J S,Cutler R A,Nacho F C,et al.Some chlorinated 4-indanols:Preparation and proof of structures[J].J Am Chem Soc, 1957, 79: 3559-61.
[10]Nakajima T,Goi T,Kawata A,et al.Pyrazolopyrimidine Compound:WO,2014030716A1[P].2014-02-27.
