高通量测序技术对常染色体隐性Alport综合征两个家系遗传特征的实验诊断研究*
2018-07-03吴维青崔英霞夏欣一刘志红李晓军
李 昂,吴维青,吕 辛,崔英霞,夏欣一,刘志红,李晓军
(1.江苏大学医学院,江苏镇江 212013;2.解放军南京总医院a.检验医学研究所;b.肾脏病研究所,南京 210002)
Ⅳ型胶原基因COL4A3,COL4A4和COL4A5突变可导致Alport综合征(Alport syndrome,AS),三个基因分别包含52,47,51个外显子,且没有突变热点,应用传统Sanger测序进行基因诊断费时费力且价格高昂[1]。COL4A3,COL4A4基因突变临床表型具有高度异质性,可导致常染色体显性AS,常染色体隐性AS,良性家族血尿(benign familial hematuria,BFH)等[2]。临床上对AS和BFH的诊疗方案及预后判断不同,疾病早期症状不典型,故很难进行鉴别诊断[3],因此,应用恰当方法对疑似AS患者及家系进行早期病因学诊断及鉴别诊断尤为重要。二代测序技术(next generation sequencing,NGS)由于其高通量、低成本等优势逐渐应用于科研及临床诊断[4]。本研究拟探讨靶基因捕获联合二代测序技术在AS病因诊断及家系不同临床表型成员遗传学实验诊断中的应用价值。
1材料与方法
1.1 研究对象 2012年1月~2014年12月期间,在南京军区南京总医院就诊的2个遗传性肾病家系,先证者依据经典gregory标准[5]确诊为Alport综合征,2家系先证者的父母均为近亲婚配,见图1。
1.2 试剂和仪器 QIAamp DNA Blood MiNi Kit(Qiagen,Hilden,Germany)基因组DNA提取试剂;课题组自行设计的遗传性肾病捕获芯片(NimbleGen,Roche);Illumina Hiseq2500测序平台(美国);ABI 3730xl DNA测序仪。
1.3 研究方法
1.3.1 基因组DNA的提取:经患者签署知情同意书后,抽取先证者及家系成员外周血4 ml,EDTA抗凝,按照全血DNA提取试剂盒标准步骤进行。
1.3.2 靶序列捕获芯片的设计:根据NCBI和OMIM数据库,确定常见遗传性肾病(包括 PKD,AS,BFH,SRNS等)的相关78个致病基因,根据这些基因序列设计特异杂交捕获探针,捕获区域包含这些基因外显子及侧翼序列,制定特异性遗传肾病相关基因捕获芯片(NimbleGen,Roche)。
1.3.3 目标区域高通量二代测序:根据标准流程进行目标序列的捕获和富集、测序文库的建立和高通量测序,大体步骤如下:使用超声波仪将基因组DNA打断变成200~300 bp的碎片(Covaris S2,Massachusetts,USA)。末端连接Illumina的寡核苷酸接头(单端),在连接完成后使用一种高保真聚合酶以及包含定制barcode序列的PCR引物4个循环的扩增来验证接头连接是否成功。通过PCR来完成文库构建并用于进一步分析,来自10个文库的DNA接头连接片段和索引片段被混在一起并与定制的寡核苷酸探针进行杂交。完成杂交和清洗后,使用300 ml洗脱缓冲液(qiagen,valencia,CA,USA) 洗脱探针溶液中与寡核苷酸探针结合在一起的DNA片段。测序引物完成杂交之后,在中国天津华大基因股份有限公司的Illumina Hiseq2500平台上(illumine,San Dieago,CA,USA)进行双端测序。
1.3.4 生物信息学分析:测序获得的原始数据由Illumina Pipeline version (1.3.4版)软件进行处理,经过滤去污染后使用BWA软件包(Burrows Wheeler Aligner)比对干净读数[6]。由GATK IndelGenotyper确定插入和缺失变异,SOAP snp软件确定SNP[7]。分析数据流程中使用的对照人群数据库包括千人基因组数据库,dbSNP数据库和华大内部数据库。采用PolyPhen-2[8]和SIFT algorithm[9]算法来预测错义突变对基因功能的影响,用Phylop (phyloP46wayPlacental)软件来计算变异位点的保守度。
1.3.5 Sanger验证:根据基因组序列,应用Primer Premier 5软件设计上述NGS测序发现的突变位点特异性引物,PCR扩增先证者及家系所有成员相应位点,应用Sanger测序法进行测序验证。
1.3.6 基因型和表型分析:综合分析2个家系所有成员的临床表型、实验室检查结果、病理诊断结果及基因突变结果。
2结果
2.1 NGS测序结果 家系F1先证者(Ⅱ1)检测到COL4A3基因纯合突变c.3769G>A(p.G1257R),家系F2先证者检测到COL4A4 基因c.1715G>C(p.G572A)纯合突变。
2.2 生物信息学分析结果 COL4A3 c.3769G>A和COL4A4 c.1715G>C均为甘氨酸置换突变,分别置换为精氨酸和丙氨酸。在千人基因组数据库,dbSNP数据库和华大内部数据库中均无频率记录,Phylop(phyloP46wayPlacental)软件预测突变高度保守,PolyPhen-2和SIFT algorithm均预测突变有害。根据ACMG标准[10],这两个错义突变均属于可能致病性突变。
2.3 Sanger测序验证结果 F1家系先证者(F1-Ⅱ1)携带COL4A3基因纯合突变 c.3769G>A(p.G1257R),先证者父母及胞妹均为此突变的杂合子,见图2;F2家系先证者(F2-Ⅱ1)携带COL4A4基因纯合突变c.1715G>C(p.G572A),先证者父母及胞妹均为此突变的杂合子,见图3。
2.4 基因型和表型分析结果
2.4.1 家系F1:具有COL4A3基因纯合突变 c.3769G>A(p.G1257R)的先证者(F1-Ⅱ1),其临床表现为血尿、蛋白尿、肌酐轻度增高伴高频感音性耳聋;电镜下基底膜厚薄不一、致密层撕裂分层;Ⅳ型胶原免疫荧光检测结果显示基底膜α3链节段缺失,α5链完全缺失;临床诊断为AS。携带杂合COL4A3 c.3769G>A突变的先证者父母(F1-Ⅰ1,F1-Ⅰ2)、胞妹(F1-Ⅱ2)的临床表现均为孤立性血尿,见表1。
家系F2:具有COL4A4 c.1715G>C(p.G572A)纯合错义突变的先证者(F2-Ⅱ1),其临床表现为血尿、蛋白尿、正常肾功能伴高频感音性耳聋;电镜下基底膜厚薄不一、致密层撕裂分层,呈“花篮样”改变;IV型胶原免疫荧光检测结果显示基底膜α3链和α5链均节段缺失;临床诊断为AS。携带COL4A4 c.1715G>C(p.G572A)杂合突变的先证者母亲(F2-Ⅰ2)及胞妹(F2-Ⅱ2)临床表现为孤立性血尿,而同样为此突变的杂合子的先证者父亲(F2-Ⅰ1)未见异常表现,见表1。

Hom,homozygote代表纯合子;Het,Heterozygote代表杂合子。

Hom,homozygote代表纯合子;Het,Heterozygote代表杂合子。
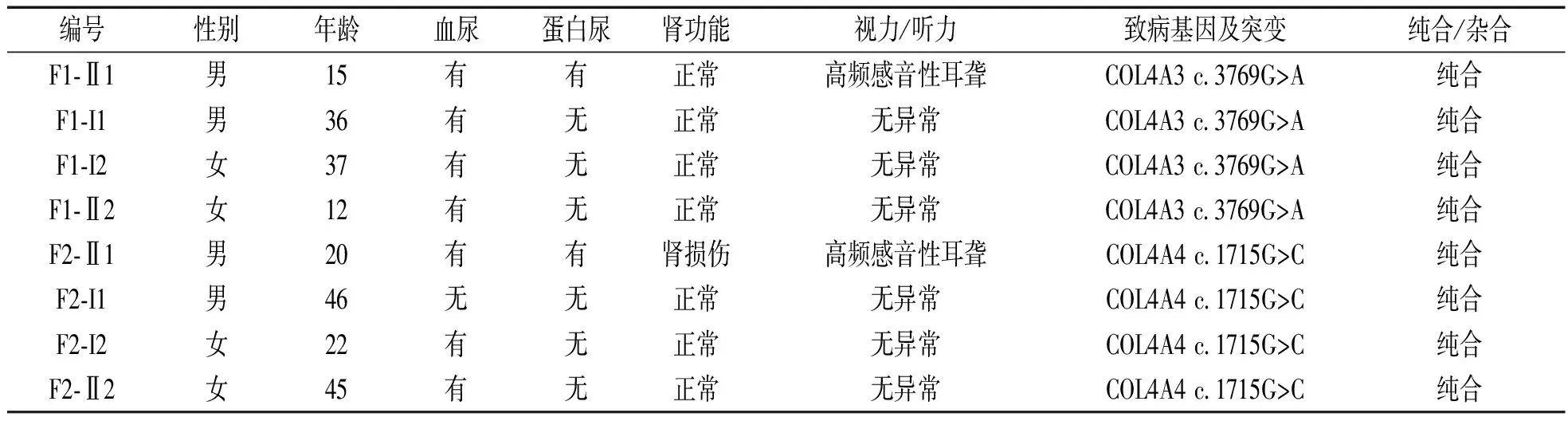
表1 两个AS家系成员临床资料和基因突变结果
3讨论Alport综合征(AS)是遗传性肾病最常见的类型之一,致病基因为Ⅳ型胶原编码基因COL4A3,COL4A4及COL4A5。COL4A3和COL4A4基因突变可导致常染色体显性或隐性Alport综合征,而COL4A5基因突变导致X连锁隐性AS[11]。这三个基因分别包含52,47,51个外显子,且没有突变热点,因而应用传统的PCR结合Sanger测序法进行致病基因突变成本昂贵且耗时耗力。本课题组,自行设计了遗传性肾病致病基因捕获芯片,结合二代测序技术,可对导致Alport综合征、多囊肾等多种遗传性肾病78个基因同时进行突变检测。我们的研究数据表明,本检测方法能够在较短时间内明确绝大多数AS患者致病原因,也可对表型较为轻微的疑似患者进行鉴别诊断。鉴于靶基因捕获二代测序技术所具有的高通量、低成本及在致病基因突变检测的有效性,在Alport综合征病因诊断、鉴别诊断等方面已经开始取代传统的Sanger测序,发挥了越来越重要的作用。
COL4A3和COL4A4基因编码Ⅳ型胶原蛋白α3,α4链,这两个基因杂合突变可引起常染色显性Alport综合征[3],薄基底膜病(thin basement membrane nephropathy,TBMB)或称为家族良性血尿(benign familial hematuria,BFH)[2],也有研究表明亦和局灶性节段性肾小球硬化症(FSGS)相关[12]。也就是说COL4A3或COL4A4基因的杂合突变表型具有非常高的异质性,携带者临床表现谱非常广泛,包含了完全正常、BFH及Alport综合征;即便是在携带同一突变的同一家系内部,不同成员的临床表现也具有明显差异。本研究家系F2中,同样为COL4A4 c.1715G>C突变携带者,F2-I 2和F2-Ⅱ1表现为BFH,而F2-I 1则完全正常,也证明了Ⅳ型胶原基因变异临床表型的异质性,这可能和疾病不全外显有关。
BFH和常染色体隐性Alport综合征可同时影响一个家系,但二者的临床表现及预后明显不同,Alport综合征临床表现为血尿、蛋白尿、进行性肾损伤伴或不伴高频感音性耳聋及视力异常,而BFH以孤立性血尿为临床特征,肾功能往往正常。早期使用血管紧张素转化酶抑制剂(ACEI)可有效缓解Alport综合征进展为终末期肾病,而BFH可仅观察及预防,所以两种疾病的早期诊断和鉴别诊断尤为重要[13~15]。以往BFH和Alport综合征的诊断依赖临床症状和病理,但在早期临床症状和病理结果不典型情况下,做出明确诊断非常困难[16],而且不是所有患者或家属能够接受活检这样的有创检测。而外周血标本进行基因诊断却具有独特的优势,相比于病理诊断,其对病人造成的创伤小且更加精准高效,结果不随病人疾病进展发生变化。本研究中,携带基因突变的夫妻生育的每个孩子都有四分之一的概率为Alport综合征患者,而两家系先证者胞妹均具有血尿表现,因而不能排除Alport综合征的可能。经过基因诊断,均被证明为基因突变携带者。我们建议,针对有血尿倾向的疑似Alport综合征的家系,尤其是有血尿家族史而无详细临床和病理资料时,家系所有成员都需进行基因诊断、明确基因型,这不仅有助于已有临床表现患者的鉴别诊断,还可以预测其他家系成员将来可能的临床表现,从而指导家系的遗传咨询和精准诊疗。
总之,我们的研究表明靶基因捕获联合高通量测序技术在Alport综合征病因诊断中发挥重要的作用;结合Sanger测序法,可对Alport综合征家系不同表型患者做出精准诊断;建议具有家族性血尿的家系所有患者都进行基于二代测序的基因突变检测,为家系的精准诊疗和遗传咨询创造条件。
参考文献:
[1] Liu JH,Wei XX,Li A,et al.Novel mutations in CO L4A3,COL4A4 and COL4A5 in Chinese patients with Alport Syndrome[J].PLoS One,2017.12(5):e0177685.
[2] Xu Y,Guo M,Dong H,et al.A novel COL4A4 mutation identified in a chinese family with thin basement membrane nephropathy[J].Sci Rep,2016,6:20244.
[3] Kamiyoshi N,Nozu K,Fu XJ,et al.Genetic,clinical and pathologic backgrounds of patients with autosomal dominant alport syndrome[J].Clin J Am Soc Nephrol,2016,11(8):1441-1449.
[4] Preston R,Stuart HM,Lennon R.Genetic testing in steroid-resistant nephrotic syndrome:why,who,when and how?[J].Pediatr Nephrol,2017,doi:10.1007/S00467-017-3838-6.(Epubahead of Print)
[5] Gregory MC,Terreros DA,Barker DF,et al.Alport syndrome-clinical phenotypes,incidence,and pathology[J].Contrib Nephrol,1996,117:1-28.
[6] Li H,Durbin R.Fast and accurate short read alignment with Burrows-Wheeler transform[J].Bioinformatics,2009,25(14):1754-1760.
[7] McKenna A,Hanna M,Banks E,et al.The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data[J].Genome Res,2010,20(9):1297-1303.
[8] Adzhubei IA,Schmidt S,Peshkin L,et al.A method and server for predicting damaging missense mutations[J].Nat Methods,2010,7(4):248-249.
[9] Ng PC,Henikoff S.Predicting deleterious amino acid substitutions[J].Genome Res,2001,11(5):863-874.
[10] Richards S,Aziz N,Bale S,et al.Standards and gui-delines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J].Genet Med,2015,17(5):405-424.
[11] Kruegel J,Rubel D,Gross O.Alport syndrome-insights from basic and clinical research[J].Nat Rev Nephrol,2013,9(3):170-178.
[12] Wu Y,Hu PZ,Xu HB,et al.A novel heterozygous COL4A4 missense mutation in a Chinese family with focal segmental glomerulosclerosis[J].J Cell Mol Med,2016,20(12):2328-2332.
[13] Kashtan C.Alport syndrome:facts and opinions[J].F1000 Res,2017,6:50.
[14] Uchida N,Kumagai N,Nozu K,et al.Early RAAS blockade exerts renoprotective effects in autosomal recessive alport syndrome[J].Tohoku J Exp Med,2016,240(3):251-257.
[15] Chew C,Lennon R.Basement membrane defects in genetic kidney diseases[J].Front Pediatr,2018,6:11.
[16] Li A,Gao EZ,Cui YX,et al.Three novel heterozygous COL4A4 mutations result in three different collagen type IV kidney disease phenotypes[J].Cytogenet Genome Res,2018,154(1):30-36.
