HPLC法测定槟榔中槟榔碱和槟榔次碱的含量
2018-06-23李春燕张学敏岳璐闫福林新乡医学院三全学院河南新乡453003新乡医学院药学院河南新乡453002
李春燕,张学敏,岳璐,闫福林*(1.新乡医学院三全学院,河南 新乡 453003;2.新乡医学院药学院,河南 新乡 453002)
槟榔为棕榈科植物槟榔ArecacatechuL. 的干燥成熟种子,广泛栽培于中国、印度、印度尼西亚、斯里兰卡、菲律宾等南亚及东南亚国家[1]。中医认为槟榔性温,味辛、苦,归胃、大肠经,具有驱虫消积、行气利水、截疟的功效,槟榔碱是槟榔中的主要活性成分,其他生物碱还有槟榔次碱、去甲基槟榔次碱等[1-3]。
目前测定槟榔碱含量的方法主要有HPLC、薄层扫描法、毛细管电泳法和酸碱滴定法等[2,4-6],而对槟榔次碱的含量测定方法尚未见文献报道。本文建立了采用反相高效液相色谱测定槟榔中槟榔碱和槟榔次碱的分析方法,该方法简便易行,灵敏可靠,可用于测定槟榔中槟榔碱和槟榔次碱的含量 。
1 仪器与试药
1.1 仪器
Agilent1100高效液相色谱仪(美国安捷伦公司);电子天平(万分之一,MS204S型,瑞士Mettler Toledo公司);电子天平(十万分之一,万分之一,XS205型,瑞士Mettler Toledo公司);微量高速离心机(TG-16W型,长沙湘仪);上海雷磁公司pH-25型pH计;德国MN Nucleosil SA阳离子交换色谱柱(4.6 mm×250 mm,5 μm)。
1.2 试药
氢溴酸槟榔碱(供含量测定用),中国药品生物制品检定所提供;盐酸槟榔次碱(供含量测定用),购于SIGMA公司;甲醇为色谱纯,水为超纯水,其余试剂为分析纯。
2 方法及结果
2.1 色谱条件
色谱柱为Nucleosil SA阳离子交换色谱柱(4.6 mm×250 mm,5 μm),流动相为甲醇-水-磷酸(60∶40∶0.3, 氨试液调pH3.8),检测波长:212 nm,流速:1.0 mL·min-1,柱温:30℃,进样量:10 μL。
2.2 溶液的制备
2.2.1 对照品溶液的制备
分别取槟榔碱和槟榔次碱对照品适量,精密称定,加40%甲醇溶解并制成每1 mL约0.5 mg的溶液,作对照品储备液,在-20℃下保存备用。分别精密量取对照品储备液适量,加40%甲醇制成每1 mL中含槟榔碱8 μg和槟榔次碱3 μg的溶液,即得。
2.2.2 供试品溶液的制备
取样品粉末约3 g,置于50 mL锥形瓶中,精密加入0.001 mol·L-1磷酸溶液30 mL,回流30 min,滤过,取1 mL滤液加入1 mL 85%磷酸,用0.45 μm滤器滤过,收集滤液,取1 mL滤液于10 mL容量瓶中加入40%甲醇溶液稀释,加入25%氨水500 μL,40%甲醇定容,摇匀,备用。
2.3 方法学考察
2.3.1 专属性试验
分别取对照品溶液和供试品溶液各10 μL注入液相色谱仪,按上述色谱条件测定,记录色谱图,结果见图1,表明其他组分对槟榔碱和槟榔次碱测定无干扰。

图1 HPLC色谱图注:A.对照品,B.供试样品;a.槟榔次碱,b.槟榔碱
2.3.2 线性关系
取槟榔碱对照品储备液,摇匀,分别精密量取0.4,0.8,1.6,2.4,4.8 mL,置100 mL棕色量瓶中,加40%甲醇稀释至刻度,摇匀,分别依法进样测定,记录色谱图,以浓度(X)为横坐标,峰面积(Y)为纵坐标,计算线性回归方程为Y=0.049 5X+1.388 1,R2=0.999 5(n=5),结果表明,槟榔碱在2.02~32.30 μg·mL-1浓度范围内呈很好的线性关系。
取槟榔次碱对照品储备液,摇匀,分别精密量取0.2,0.4,0.8,1.6,2.4 mL,置100 mL棕色量瓶中,加40%甲醇稀释至刻度,摇匀,分别依法进样测定,记录色谱图,以浓度(X)为横坐标,峰面积(Y)为纵坐标,计算线性回归方程为Y=0.045 1X+0.128 3,R2=0.999 3(n=5),结果表明,槟榔次碱在0.85~13.55 μg·mL-1浓度范围内呈很好的线性关系。
2.3.3 精密度试验
精密量取对照品储备液适量,制成每1 mL中约含槟榔碱8 μg和槟榔次碱3 μg的对照品溶液,分别连续进样6次,槟榔碱和槟榔次碱峰面积的RSD依次为0.48%和0.67%,结果表明,本法进样的精密度良好。
2.3.4 重复性试验
取同一样品,按“3.2供试品溶液的制备”项下方法,平行制备6份供试品溶液,依次进样,6份供试品中槟榔碱和槟榔次碱峰含量的RSD为2.79%和3.01%。结果表明,本法的重复性良好。
2.3.5 溶液稳定性试验
取对照品溶液和供试品溶液,室温下放置0,2,4,8,12,24 h,分别进样,槟榔碱和槟榔次碱峰对照品溶液峰面积的RSD为1.87%和2.31%,供试品溶液中槟榔碱和槟榔次碱峰峰面积的RSD为2.18%和2.74%,结果表明,对照品溶液和供试品溶液24 h内稳定性较好,满足测定要求。
2.3.6 加样回收试验
精密量取槟榔碱和槟榔次碱对照品储备液适量,加40%甲醇定量稀释制成每1 mL中约含槟榔碱100 μg和槟榔次碱50 μg的工作溶液。取已测得含量的供试品溶液9份,精密称定,精密加入0.5,1.0,1.5 mL上述工作溶液,按照含量测定法测定。槟榔碱的平均回收率为97.2%,RSD为2.4%(n=9);槟榔次碱的平均回收率为103.9%,RSD为2.6%(n=9)。
2.4 样品测定
取不同产地的10批样品,照上述测定方法和色谱条件依法测定,样品中槟榔碱和槟榔次碱含量测定结果如表1。
3 讨论与结论
3.1 色谱条件的筛选
参照相关文献[4,6],选择检测波长为412 nm。分别比较了不同色谱柱Diamonsil C18色谱柱和Nucleosil SA阳离子交换色谱柱,并考察了流动相配比以及柱温为30℃、35℃和40℃时的分离情况,在满足槟榔碱和槟榔次碱与杂质峰基线分离的前提下,并使待测物质色谱峰峰形良好的情况下,最好选择色谱条件为色谱柱为Nucleosil SA阳离子交换色谱柱(4.6 mm×250 mm,5 μm),流动相为甲醇-水-磷酸(60∶40∶0.3, 氨试液调pH3.8),检测波长:212 nm,流速:1.0 mL·min-1,柱温:30℃。
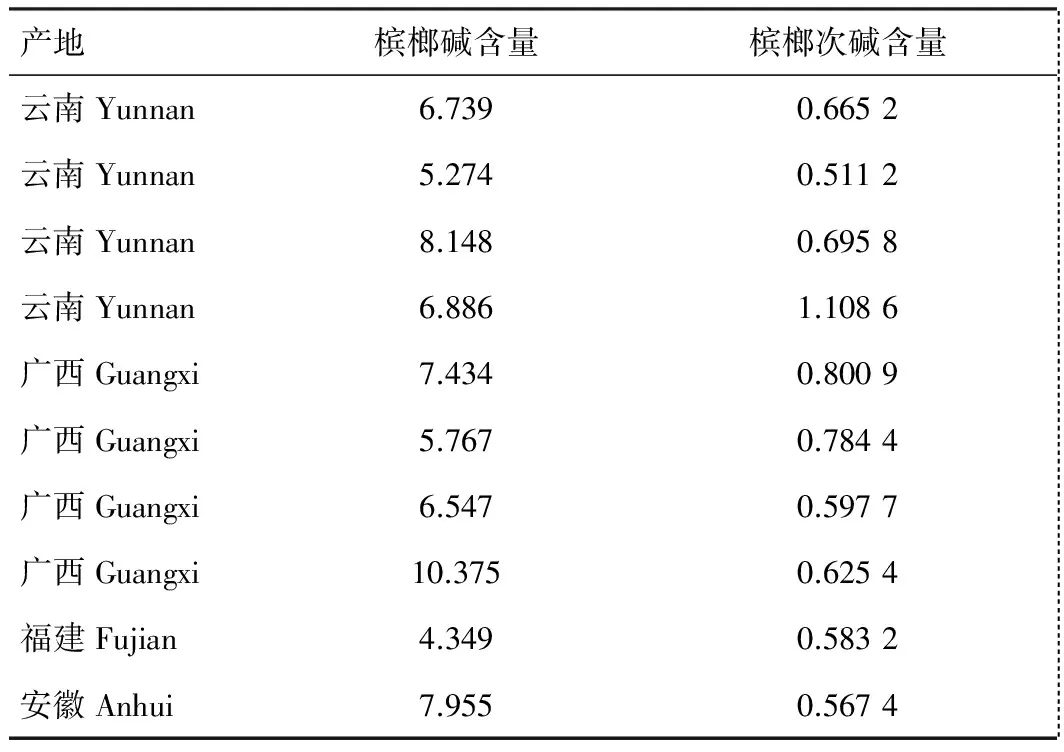
表1 样品中槟榔碱和槟榔次碱含量测定结果(mg/g)
3.2 样品前处理方法的筛选
分别比较了不同溶剂体系:(1)甲醇,(2)40%甲醇,(3)20%甲醇,以及回流时间20 min、30 min和60 min,结果表明采用回流30 min的提取效果良好,可以用于本方法体系的含量测定。
3.3 槟榔产地分析
本文所分析的10批槟榔样品中,槟榔碱含量均高于0.20%,符合2015版中国药典槟榔项下的要求,其中除安徽产的槟榔中槟榔碱含量稍低外,其他产地的槟榔中槟榔碱的含量无显著性差异。由于槟榔次碱也是槟榔中的重要活性成分之一,本方法同时测定了槟榔次碱的含量,在10批槟榔样品中槟榔次碱的含量均高于0.05%,故建议药典中应列入槟榔次碱的含量测定项或相关质量控制项,已便更好的控制槟榔质量。
综上所述,本文所建方法重复性好,回收率高,能准确测定槟榔中槟榔碱和槟榔次碱的含量,为槟榔质量控制和质量标准的完善提供了参考。
[1] 李习雄,胡冠英,张三印.槟榔毒性机制的研究进展[J].中国实验方剂学杂志,2015,21(19):212-216.
[2] 付克祥,周轶新.槟榔检测技术的研究与应用[J].农业技术与装备,2015,10(10):13-15.
[3] 杨文强,王红程,王文婧,等.槟榔化学成分研究[J].中药材,2012,35(3):400-403.
[4] 杨华,郭元满,谭泽云,等.HPLC法同时测定四磨汤滴丸中辛弗林、槟榔碱和去甲异波尔定的含量[J].中医药导报,2016,22(2):40-43.
[5] 伦志彩,隋莹,徐刚,等.正交试验优化槟榔免煎颗粒提取工艺[J].中国医药导报,2016,13(1):43-45,53.
[6] 马玉红.槟榔碱的含量测定[J].中国民族民间医药,2013,22(17):12,14.
