ω-转氨酶不对称合成手性胺及非天然氨基酸的研究进展
2018-06-12王亚军
程 峰,相 超,王亚军
(1.浙江工业大学 生物工程学院,浙江 杭州 310014;2.浙江工业大学 浙江省生物有机合成技术研究重点实验室,浙江 杭州 310014)
手性胺是指小分子化合物手性中心含有氨基的一类化合物,是众多医药及农药的关键中间体,在国民经济中占据重要地位[1-2]。例如,糖尿病类治疗药物——西他列汀(Merck公司及Codexis公司)及广谱触杀型除草剂——草铵膦(Bayer公司)都具有手性胺化学模块。
目前,化学法、生物拆分法和生物不对称合成法是制备手性胺的主要方法。化学法合成手性胺往往需要使用昂贵的金属催化剂及特制的溶剂,生产成本较高,对映选择性较低,并会造成一定的环境污染;生物催化剂进行手性拆分法制备高纯度手性胺的最大理论收率只有50%(图1(a))[3]。因此,这两种方法都不能满足实际生产的需要。生物法生产手性胺的首选策略为不对称合成,其理论收率高达100%(图1(b)),使其在生产工业领域有着强大的应用前景,相关研究受到了广泛的重视。针对氨基转移可逆的特点,通过一些策略可以将反应平衡向胺生成的方向移动,能够显著提高手性胺的生产效率。
本文中,笔者介绍ω-转氨酶的结构特征,并以制备西他列汀中间体等为例,阐述了ω-转氨酶的高通量筛选方法及分子改造的研究进展及通过级联反应提高手性胺产量的策略。最后,综述了ω-转氨酶在不对称合成非天然氨基酸中的具体应用。
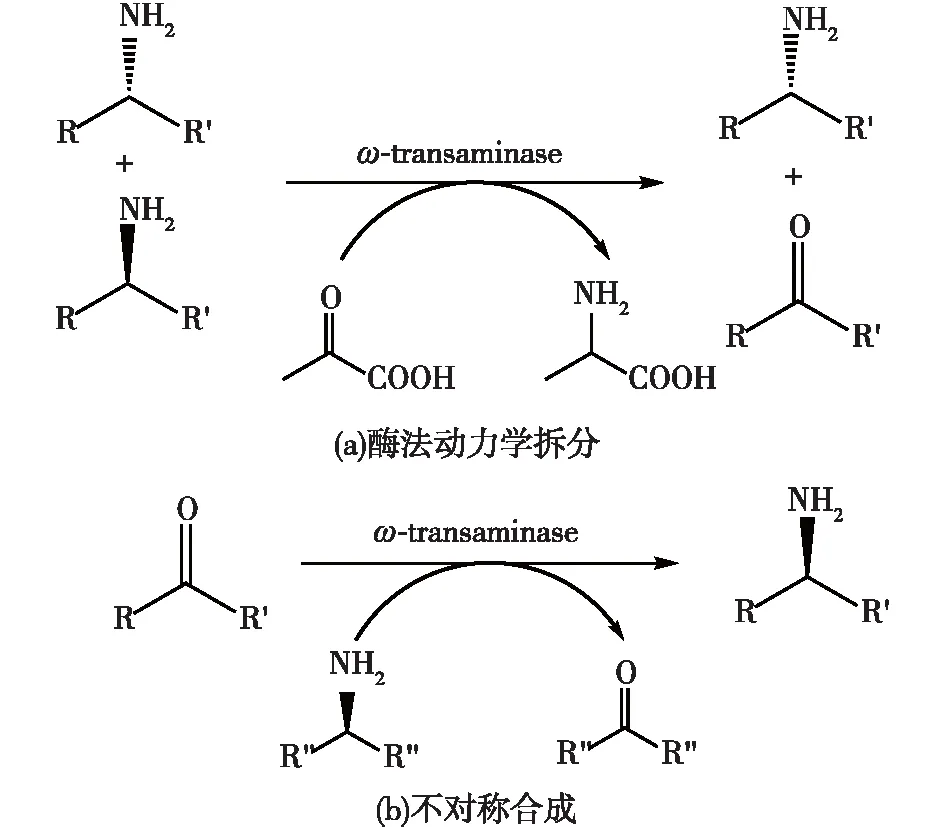
图1 酶法动力学拆分(a)与不对称合成(b)制备手性胺的基本原理Fig.1 Principles of chiral amines synthesis by kinetic bio-resolution (a) and asymmetric synthesis (b)
1 ω-转氨酶的结构特点及催化机制
转氨酶(transaminase,TA,EC 2.6.1.X),又称氨基转移酶(aminotransferase),属于转移酶类,能够可逆催化酮基与氨基之间的氨基转移反应。当这类酶催化的转氨反应中,底物或产物含有α-氨基酸时,就称该酶为α-转氨酶,反之则称之为ω-转氨酶[1]。α-转氨酶的产物一般只是α-氨基酸,而ω-转氨酶能够氨基化酮酸、醛和酮,且具有立体选择性高、辅因子可再生的特点,因此,ω-转氨酶被更广泛地应用于合成医药和农药中间体[4]。
1.1 ω-转氨酶的三维结构特征
ω-转氨酶为磷酸吡哆醛(PLP)依赖型酶,通过不对称催化还原胺酮合成手性胺,通常使用丙氨酸或异丙胺作为氨基供体。根据一般规律,使用D-丙氨酸作为氨基受体的ω-转氨酶为(R)-型转氨酶,属于Ⅳ类折叠(图2(a));而使用L-丙氨酸为氨基供体的ω-转氨酶为(S)-型转氨酶,属于Ⅰ类折叠(图2(b))。这两类折叠的ω-转氨酶有着不同的序列长度、不同的折叠方式,缺乏整体序列相似性,因此,它们的进化是分开进行的[5]。
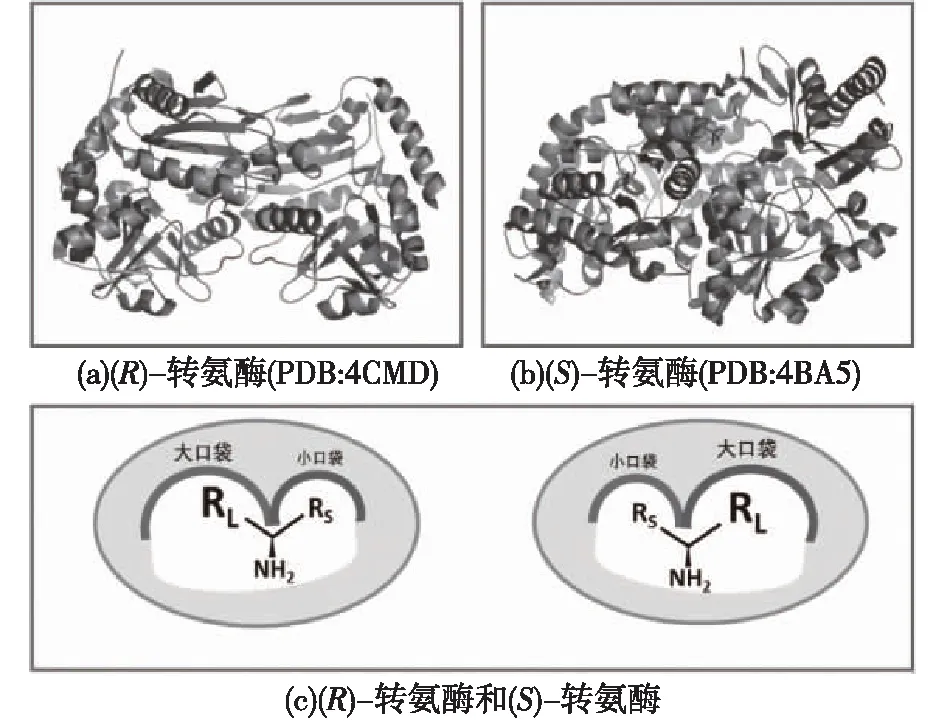
图2 (R)-型转氨酶的三维结构一般特征(a)(PDB:4CMD),(S)-型转氨酶的三维结构一般特征(b) (PDB:4BA5)及两种转氨酶底物结合口袋的一般结构(c)Fig.2 3D structure of (R)-selective transaminase(a)(PDB:4CMD),(S)-selective transaminase(b)(PDB:4BA5),and the structural feature of active sites in (R)- and (S)-transaminases(c)
尽管这两类转氨酶有以上的不同特征,但是双底物识别是它们的一个共同特点。所有已知的野生型TAs的活性中心都由1个大口袋和1个小口袋组成,亲水和疏水基团的底物都可以结合到转氨酶两个口袋中(图2(c))。在两个口袋中,这两类ω-转氨酶的关键催化氨基酸残基有着相同的空间排列,而且都具有高度保守的赖氨酸残基。有研究者对一些(S)-TAs结构和(R)-TAs结构进行了分析,提出了关于TAs在活性位点处可接受疏水性和亲水性底物的相关理论[6]。研究者认为,(S)-转氨酶通过“翻转”活性位点附近的一个“环”结构上的精氨酸残基,对活性部位的胍基部分进行移动,使底物的羧基部分可以进入活性口袋进行反应;在需要疏水环境时,将胍基部分移动出活性部位以提供疏水环境,这一过程在双底物识别过程中发挥了关键作用。例如,来自Chromobacteriumviolaceum的(S)-TA的Arg129位点[7]。而对于(R)-TA,同样是一个位于活性部位附近的精氨酸残基显示类似的功能:Aspergillusfumigatus中(R)-TA的Arg126位点和Aspergillusterreus中(R)-TA的Arg128位点[8]。但是,目前已发现的大部分野生型ω-转氨酶的小口袋只能容纳1个甲基[1],这严重影响了其对大体积底物的催化活性。
1.2 ω-转氨酶的基本催化机制
针对ω-转氨酶双底物识别的特征,目前公认的反应机制主要包括两步半反应的Ping-Pong Bi-Bi机制(图3)。在第一个半反应中,酶与PLP结合(E:PLP)形成内部醛亚胺结构,同时催化中心的赖氨酸亲核攻击氨基供体产生外部醛亚胺结构;接着,中性赖氨酸残基释放,平面醌类中间体催化内部氢重排,产生酮亚胺。在第二个半反应中,酮亚胺水解,释放产物酮并产生复合体酶:吡哆胺5′-磷酸(E:PMP);然后,在E:PMP和底物酮之间形成酮亚胺络合物,其最终经由第二平面醌型中间体和外部醛亚胺再生E:PLP,并最终释放出产物——手性胺[1,9]。
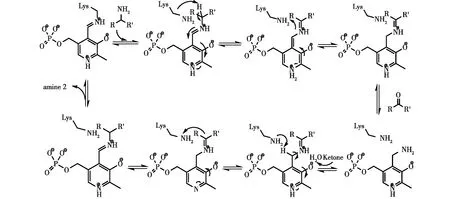
图3 ω-转氨酶催化不对称合成手性胺的双底物乒乓机制Fig.3 Principles of asymmetric synthesis of chiral amines by ω-transaminase(Ping-Pong Bi-Bi mechanism)
2 ω-转氨酶的筛选及改造
酶法制备光学纯手性胺的关键在于获得高活力与高选择性兼备的ω-转氨酶。但由于ω-转氨酶种类相对偏少,底物谱窄,因此,通过筛选或改造得到性质优良的ω-转氨酶非常重要。近年来,许多研究集中在筛选新型ω-转氨酶,通过改造ω-转氨酶关键结构域增大其底物谱等方面,而且这些研究也取得了一定的突破[1-2]。
2.1 ω-转氨酶的筛选
ω-转氨酶的筛选方法主要分为传统的微生物培养筛选方法和计算机筛选方法。前者首先需要对微生物进行富集培养,进而通过使用唯一氮源筛选得到活性以及选择性强的转氨酶。目前,通过该方法发现的ω-转氨酶序列具有较大差异,如使用(S)-α-甲基苄基胺为唯一氮源筛选得到KlebsiellapneumoniaJS2F、BacillusthuringiensisJS64及VibriofluvialisJS17等菌株。从这些菌株中克隆得到的ω-转氨酶氨基酸序列一致性小于50%,可以说明ω-转氨酶来源较为广泛。而且这种筛选方法在实际操作过程中大多以L-丙氨酸为氨基供体,得到的ω-转氨酶绝大多数是(S)-选择性[4]。尽管大量的努力来筛选,但是目前只有少数几个ω-转氨酶是R-选择性,如来自Aspergillusterreus菌的ω-转氨酶(PDB:4CE5)[10]。因此,传统筛选难以快速获得(R)-选择性的ω-转氨酶。
针对这一问题,有研究者开发出计算机虚拟筛选(R)-选择性的ω-转氨酶。计算机筛选方法是使用生物信息学和计算机等手段在已有的数据库中对酶进行筛选和分析,通过序列对比、建模对酶活性位点对比分析以及基序搜索等方法,从基因序列中筛选可能具有活性的酶基因。2010年,德国Bornscheuer教授课题组的Höhne等[3]成功开发出一种基于序列-底物特异性和立体选择性的程序,预测功能未知的(R)型选择性ω-转氨酶。首先,他们通过替换活性位点关键的氨基酸残基对蛋白质活性位点进行合理设计并且预测蛋白质活性。然后,通过数据库对预测蛋白进行搜索并获得携带突变的相似蛋白,这样可以充分利用经过数百万年的自然进化选择的蛋白质而非重新设计蛋白。使用这种策略,他们成功从5 700个序列中鉴定出21个(R)-选择性的TA。其中,17个ω-转氨酶在验证实验中显示出(R)-选择性的转氨活力。最终,7个(R)-ω-转氨酶被成功应用于合成 (R)-的脂肪族和芳香族类的手性胺。类似地,Iglesias等[11]利用这种基因挖掘的方法,成功获得了来源于Caproniasemiimmersa的(R)-ω-转氨酶,当以苄基丙酮或乙酰苯作为底物时,该酶显示了很好的立体选择性(对于这两个底物,该酶的对映体选择率(E)>200,产物的对映体过量值e.e.>99%)。
2.2 ω-转氨酶改造
针对天然的ω-转氨酶底物谱较窄及其立体选择性单一(基本是(S)-型)的特点,近年来,有较多关于ω-转氨酶改造的报道,特别在酶的立体选择性及底物特异性方面。应用改造后的转氨酶获得与医药中间体模块的大型手性胺和(R)型立体选择性手性胺,这是一种非常具有前景的合成方法[12]。
2.2.1 改造ω-转氨酶改变其立体选择性
目前已经发现了大量编码ω-转氨酶的基因,这些编码基因的发现对ω-转氨酶合成不同的手性胺起到了巨大的推动作用[2]。最著名的改造ω-转氨酶立体选择性的例子是2011年美国Codexis公司对来自Arthrobactersp.的ATA117进行的改造。在引入27个突变点后成功得到ATA117-Rd11这一突变蛋白,其立体选择性改变为严格的(R)型。该突变体酶能够催化西他列汀前体酮不对称合成糖尿病类治疗药物西他列汀(一种R-构型手性胺)[13]。改造中的涉及27个突变主要集中在3个结构区域: ①底物结合区域(包括大口袋或者小口袋);②辅酶PLP结合部位;③129~145的“环”结构[6]。Codexis公司利用多轮改造后的ω-转氨酶,成功实现了R型选择性的不对称转氨过程,实现了西他列汀的一步不对称合成。
2.2.2 改造ω-转氨酶提高其对大型底物的催化活性
ω-转氨酶本身结构特征使天然的ω-转氨酶很难催化大底物进行反应,一般认为主要是由于活性口袋空间位阻影响大底物进入活性中心导致。但是,随着近年来通过理性设计突变加强ω-转氨酶对大底物活性研究的时有报道,如Pavlidis等[14]研究指出:位阻并不是影响大底物反应进行的唯一因素,反应物在特定位点绑定并相互作用是提高催化效率的一个关键因素;他们同时发现,改变ω-转氨酶的选择性可能需要重新构建酶的整个底物口袋。同样是上述例子的ATA117,Savile等[13]利用蛋白质工程改造后,使得野生型的底物结合口袋体积充分扩大,使突变体酶能够接受西他列汀前体酮作为底物;而野生型的ATA117由于底物结合口袋的限制,不能结合西他列汀前体酮。在改造中所涉及的突变主要集中在:①酶底物结合大口袋(S223P显著增加了大口袋结合比苯环大的基团的能力);②底物结合小口袋(V69G、F112I和A284G使小口袋能够接受比甲基大的基团);③129~145的“环”结构(“环”结构上的突变降低了“环”旋转时造成的空间位阻)[6]。利用多轮改造后的ω-转氨酶,Codexis公司成功实现了R型选择性的转氨过程,并解决了工业生产反应中高浓度的有机溶剂、高浓度氨基供体(异丙胺)以及产品积累的问题。同时,值得注意的是,在扩大口袋和提高活性的改造过程中,转氨酶ATA117的高选择性却没有降低。
现将(S)-选择性和(R)-选择性的ω-转氨酶及其催化合成手性胺的研究进行总结,具体见表 1~2。

表1 已知的(S)-选择性ω-转氨酶及其催化合成的手性胺种类
2.3 ω-转氨酶高通量筛选策略的构建
ω-转氨酶的改造过程中需要一种的稳定、灵敏和高通量的筛选方法,现在主要通过高效液相色谱(HPLC)检测,或建立一种酶活检测来筛选[24]。由于ω-转氨酶的不断发现及其在合成高价值化合物中的应用发展迅速,近几十年来对ω-转氨酶高通量筛选方法的发展也随之加快。基于通用底物的高通量检测方法是首选方案[25],因为它可以更快地产生结果,并且可以快速开始检测[26]。但是,ω-转氨酶的工业应用底物大多为非天然化合物,使得高通量筛选得到的有益突变体不一定会对目标底物具有较高的活性,所以往往在高通量筛选之后还需要HPLC进行复测[19]。

表2 已知的(R)-选择性ω-转氨酶及其催化合成的手性胺种类
目前的检测手段主要包括紫外(UV)、pH、导热率、显色及荧光等技术(图4~5)[22]。大多数TA催化的产物或者底物中会有酮酸或氨基酸,因此,转氨酶早期的筛选方法主要基于毛细管电泳[27],通过离子交换液相色谱法或分光光度检测器直接检测产生的氨基酸。此外,将转氨过程与脱氢酶作用偶联,可以在340 nm处对烟酰胺腺嘌呤二核苷酸磷酸(NADPH)辅因子进行分光光度监测[28-29],达到筛选的目的。多种显色物质已被设计用于转氨酶测定,例如一种偶联酶测定法,该测定法允许使用丙酮酸作为胺受体筛选酶和胺底物,主要的转氨反应通过L-或D-氨基酸氧化酶和辣根过氧化物酶偶联,所得的H2O2与连苯三酚红组合检测[22]。Weiss等[30]基于类似的转氨酶/氨基酸氧化酶原理,建立了使用乙醛酸盐作为胺受体的基于甘氨酸-氧化酶的测定法,该技术已成功地应用于(R)-和(S)-选择性转氨酶的筛选。

图4 基于UV、pH和电导率检测ω-转氨酶酶活的高通量筛选方法Fig.4 High-throughput screening methods employing UV (a,b),pH (c) and conductometric analysis (d) for ω-transaminase activity detection
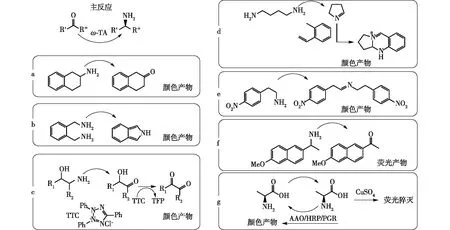
图5 比色法和荧光法进行高通量筛选Fig.5 High-throughput screening methods employing colorimetric and fluorometric analysis
四唑盐偶联显色法是另一种常用的检测手段。当四唑红被还原时,α-羟基酮被氧化为二元酮或醛酮,形成红色沉淀,使用微孔板进行读数检测。这种方法灵敏度高并且背景干扰度低,可用于ω-转氨酶对一些氨基醇胺类底物改造过程时的快速检测。但是,这种方法无法用于检测ω-转氨酶的对映体选择性[31]。通过ω-转氨酶反应过程中pH的变化进行显色反应则是另一种检测手段,一般利用甲基红作为指示剂[32]。但是这种方法灵敏度低,对于低活性的ω-转氨酶检测比较困难。类似地,有许多利用显色反应来快速检测ω-转氨酶活性的方法,如使用CuSO4/甲醇与生成的α-氨基酸生成蓝色络合物[33],采用紫外分光光度测定法测定络合物在595 nm处的吸光度,用以检测ω-转氨酶的活性[25]。
Scheidt等[34]报道了一种新的ω-转氨酶活性测定的方法。该方法使用ω-转氨酶转胺基后形成6-甲氧基萘-2-羰基荧光素,它能产生蓝色荧光。这种方法允许对底物部分基团进行替换,有助于在ω-转氨酶蛋白改造过程中对小口袋和大口袋的大小进行初步的检测。根据酮类化合物的荧光光谱,在330(λmax=310 nm)和460 nm(λmax=450 nm) 的荧光强度下,酮化合物浓度≥ 6 μmol/L的条件下,荧光强度与酮浓度之间成线性关系。该方法特点是反应物用量少而且具有很高的灵敏度,方法简单和底物消耗量低,非常适合用于对酶进行定向进化中的活性检测,特别是在ω-转氨酶的L口袋及S口袋容纳不同基团的研究过程中。
3 级联反应提高手性胺产量
鉴于ω-转氨酶催化的氨基转移反应属于可逆反应,为了使反应充分向手性胺合成的方向进行,通常有两种方法,达到此目的。一种方法是在单一酶催化系统中使用过量的氨基供体(如,异丙胺)来驱动转氨基反应,推动反应尽可能朝一个方向进行[1]。这种方法只需要单一的ω-转氨酶,在不对称手性胺合成中最简单且应用最广。但这种方法的缺点是产生的物质即为逆反应的底物从而抑制正向反应进行,因此需要加入大量胺基供体催化反应向胺生产的方向进行。这种方法往往需要联合物理方法(如,降低压力、提高温度等)移除副产物[35-36]。
第二种方法是进行多酶偶联催化反应,ω-转氨酶的偶联催化反应一般用于消除底物抑制或者进行多级反应[22,37]。ω-转氨酶催化转胺反应中生成的副产物会抑制反应的正向进行。所以,在转胺反应中,如何去除胺基供体的副产物非常重要[38]。所以在非对称合成大型手性胺中,往往会根据所使用的胺基供体选择性移除副产物[39-41]。例如,利用乳酸脱氢酶将抑制剂丙酮酸从反应中移除,将反应平衡向正方向推动[37],实现胺供体再循环;或者,加入普通催化量的胺基供体,使用氨基酸脱氢酶催化NH3再生成胺基供体,推动反应向正方向进行[12]。
Kroutil等[39]和Simon等[40]用氨基酸脱氢酶将丙酮酸再生为丙氨酸,丙氨酸可以再次用作胺基供体,因此反应体系中只需加入少量的丙氨酸,可降低副产物丙酮酸的影响。另外,为了保持氨基酸脱氢酶的活性,同时使用葡萄糖脱氢酶再生氨基酸脱氢酶的辅酶NADH,使反应持续向正方向进行,提高手性胺产量(图6)[23]。
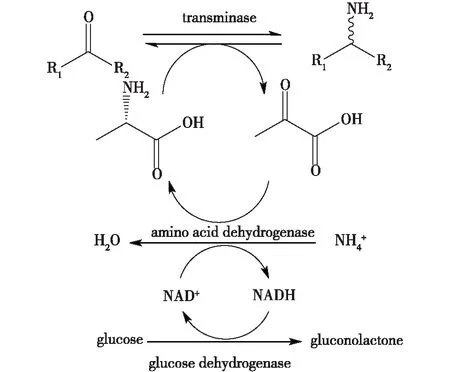
图6 转氨酶偶联氨基酸脱氢酶的反应方程式Fig.6 The cascade reaction coupling transaminase and amino acid dehydrogenase
Truppo等[37]开发了一种通过将ω-转氨酶与氨基酸氧化酶级联反应解除反应抑制提高ω-转氨酶催化合成手性胺的方法,可用于筛选立体选择性优良的ω-转氨酶。该方法将胺基底物和丙酮酸在ω-转氨酶的催化下进行反应,通过添加氨基酸氧化酶利用氧分子将丙酮酸转化为D-丙氨酸或L-丙氨酸,消除产物抑制,促进正向反应的进行。丙氨酸被氨基酸还原酶催化与O2发生不可逆反应生成H2O2,H2O2的产生可以通过添加焦糖红和辣根过氧化物酶进行检测[22](图7)。

图7 转氨酶偶联氨基酸氧化的反应方程式Fig.7 The cascade reaction coupling transaminase and amino acid oxidase
France等[42]使用羧酸还原酶(CAR),亚胺还原酶(IRED)结合ω-转氨酶的一锅法催化合成单取代和双取代的哌啶和吡咯烷,获得光学纯手性单取代-双取代的哌啶和吡咯烷(图8)。使用生物级联催化,反应从酮酸或酮醛开始,取代生成哌啶或吡咯烷骨架。在保持高催化效率的同时表现出高区域选择性及立体选择性。他们还系统研究了影响一个甲基环上取代基的位置被IRED催化减少手性亚胺,在反应过程中观察到反应过程中各种底物与不同酶之间会形成一种平衡,反应中使用来自Mycobacteriummarinum的羧酸还原酶可选择性地将羧酸转化为醛类,再使用ω-转氨酶ATA113或ATA117选择性合成不同构型的手性中心,最后使用(R)- 或(S)-IRED合成双手性中心的哌啶和吡咯烷。这种方法成功地实现了使用多酶偶联控制催化合成双手性中心的哌啶和吡咯烷[43](图9)。

图8 生物法合成2,x-二取代基吡啶(n=0)或吡咯(n=1)路线Fig.8 The biosynthesis route of 2,x-pyridine (n=0)or pyrrole (n=1)

图9 生物法合成2,6-二取代基吡啶或2,5-二取代基吡咯的路线Fig.9 The biosynthesis route of 2,6-pyridine or 2,5-pyrrole
4 ω-转氨酶不对称合成手性胺及非天然氨基酸
由于制药、化妆品、食品、化学和农业等不同行业的需求,非天然氨基酸的工业化生产日渐重要。通常,采用直接提取或者发酵法生产氨基酸,但是,对于非天然氨基酸,使用发酵法生产目前尚未见工业化的例子。目前,ω-TA已经越来越多地被应用于不对称合成生产α-和β-非天然氨基酸,这种通过酶催化方法在合成光学纯手性胺方面显示出了一定的潜力。
4.1 ω-转氨酶制备α-非天然氨基酸
以制备L-高丙氨酸为例,光学纯L-高丙氨酸可由ω-TA将2-氧代丁酸和苄胺不对称催化合成[44]。然而,反应过程中产生的苯甲醛会造成严重的产物抑制,导致转化率急剧降低。为减轻对苯甲醛的产物抑制,引入了己烷作为反应溶剂的双相反应体系。双相体系能够很大幅度地减少产物抑制现象,使产率从39%增加到96%,e.e.>99%。L-高丙氨酸也可以通过从L-苏氨酸进行生产,反应主要通过苏氨酸脱氨酶(TD)和ω-TA组成酶偶联反应。在此反应中,TD将L-苏氨酸脱氨成2-氧代丁酸,再通过ω-TA不对称转化成L-高丙氨酸。该体系的主要优点在于避免了以α-酮酸作为反应物,从而可以促进反应进行[46]。Bea等[46]通过D-氨基酸氧化酶(D-AAO)和ω-TA联合反应,使用去醛化方法从外消旋-高丙氨酸生成光学纯的L-高丙氨酸,该方法主要采用双相反应体系,使用全细胞反应将500 mmol/L外消旋-高丙氨酸转化为485 mmol/L 光学纯L-高丙氨酸 (e.e.>99%),而且该ω-TA也可以用于从3-氟丙酮酸和(S)-α-甲基苄胺不对称催化合成(R)-3-氟丙氨酸,转化率为95%,e.e.>99%[47]。
因为α-TA催化的反应平衡常数较低,光学纯的(S)-和(R)-氨基酸可以使用偶联(α/ω)-TA反应进行合成,以提高光学纯氨基酸和手性胺的收率。如,(S)-苯丙氨酸、(S)-高苯丙氨酸和(S)-天冬氨酸等(S)-型氨基酸的生产主要通过AlaTA/ω-TA、TyrTA/ω-TA进行偶联反应[48]。然而,由于ω-TA反应产生的副产物酮在高浓度下会产生严重的产物抑制,导致产率过低。因此,与生产L-高丙氨酸类似,可引入双相反应体系克服产物抑制,提高产物收率。通过分析有机溶剂分配系数,及其对酶活影响和生物相容性,最终选择邻苯二甲酸二辛酯-水作为双相反应体系,最终,不对称合成(S)-苯丙氨酸和L-高丙氨酸,的转化率分别达到93%和95%[50]。同时,该反应体系被应用于(R)-α-甲基苄胺的生产,转化率分别为56%(e.e.>95%)和54%(e.e.>96%)。另外,脂肪族的(S)-氨基酸,如(S)-缬氨酸和(R)-亮氨酸也可用同样的体系生产[48]。
4.2 ω-转氨酶制备β-非天然氨基酸
β-氨基酸主要用于制备抗生素,酶抑制剂等具有药理学性质的化合物。如,具有抗真菌和抗肿瘤活性的顺戊霉素和紫杉醇就是通过β-氨基酸合成的[49]。目前,ω-TA已成功用于生成脂肪族和芳香族β-氨基酸。例如光学纯的脂肪族β-氨基酸(D-β-氨基-正丁酸)可以通过偶联来自Mesorhizobiumsp.的β-TA与脂肪酶,针对酮羧酸酯底物不对称合成光学纯的芳族β-氨基酸,收率>20%,e.e.> 99%。尽管这种酶被命名为β-TA并且强调其对β-氨基酸的活性,但是该酶通常可以被称为ω-TA。在之后的研究中,该酶的蛋白质序列被鉴定为ω-TA类蛋白序列[50]。
5 结语
随着手性药物的发展,手性胺类化合物作为药物中间体变得越来越广泛。ω-转氨酶作为一种重要的工业用酶,主要用于不对称合成手性胺类药物中间体,其催化的反应作为合成手性胺类药物在提高产品光学纯度、保护环境等方面有着非常重要的意义,相关研究变得越来越重要。但是目前能够应用于工业应用的ω-转氨酶较少,其原因主要是:1)大部分ω-转氨酶序列相似度较低,使ω-转氨酶的研究还需要大量实验来验证;2)天然ω-转氨酶本身活性口袋较小,使得相当一部分ω-转氨酶不能满足实际应用;3)绝大多数天然ω-转氨酶是S-选择性,自然界缺乏R-选择性的ω-转氨酶。
因此,挖掘新的ω-转氨酶基因、改造现有的ω-转氨酶以及探究ω-转氨酶的反应特点变得尤其重要。今后ω-转氨酶的研究主要会集中在ω-转氨酶新酶的筛选和改造、级联反应提高手性胺的产率上,最终实现ω-转氨酶对不同大小、不同构型手性胺的不对称合成。
参考文献:
[1] MATHEW S,YUN H.ω-Transaminases for the production of optically pure amines and unnatural amino acids[J].ACS Catal,2012,2(6):993-1001.
[2] KOSZELEWSKI D,TAUBER K,FABER K,et al.ω-Transaminases for the synthesis of non-racemic alpha-chiral primary amines[J].Trends Biotechnol,2010,28(6):324-332.
[4] GUO F,BERGLUND P.Transaminase biocatalysis:optimization and application[J].Green Chem,2017,19(2):333-360.
[5] BUSS O,BUCHHOLZ P C F,GRAFF M,et al.Theω-transaminase engineering database (oTAED):a navigation tool in protein sequence and structure space[J].Proteins,2018,5(86):566-580.
[6] GUAN L J,OHTSUKA J,OKAI M,et al.A new target region for changing the substrate specificity of amine transaminases[J].Sci Rep,2015,5:2045-2322.
[7] SAYER C,ISUPOV M N,WESTLAKE A,et al.Structural studies ofPseudomonasandChromobacteriumω-aminotransferases provide insights into their differing substrate specificity[J].Acta Crystallogr D Biol Crystallogr,2013,69(4):564-576.
[8] THOMSEN M,SKALDEN L,PALM G J,et al.Crystallographic characterization of the (R)-selective amine transaminase fromAspergillusfumigatus[J].Acta Crystallogr D Biol Crystallogr,2014,70(4):1086-1093.
[9] SHIN J S,IM B G.Kinetic modeling ofω-transamination for enzymatic kinetic resolution ofα-methylbenzylamine[J].Biotechnol Bioeng,1998,60(5):534-540.
[10] LYSKOWSKI A,GRUBER C,STEINKELLNER G,et al.Crystal structure of an (R)-selectiveω-transaminase fromAspergillusterreus[J].PloS ONE,2014,9(1):e87350.
[11] IGLESIAS C,PANIZZA P,GIORDANO S R.Identification,expression and characterization of anR-ω-transaminase fromCaproniasemiimmersa[J].Appl Microbiol Biotechnol,2017,101(14):5677-5687.
[12] FUCHS M,FARNBERGER J E,KROUTIL W.The industrial age of biocatalytic transamination[J].Eur J Org Chem,2015(32):6965-6982.
[13] SAVILE C K,JANEY J M,MUNDORFF E C,et al.Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture[J].Science,2010,329:305-309.
[14] PAVLIDIS I V,WEISS M S,GENZ M,et al.Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines[J].Nat Chem,2016,8(11):1076-1082.
[15] SHIN J S,YUN H,JANG J W,et al.Purification,characterization,and molecular cloning of a novel amine:pyruvate transaminase fromVibriofluvialisJS17[J].Appl Microbiol Biotechnol,2003,61(5/6):463-471.
[16] HWANG B Y,KIM B G,KIM J H.Bacterial surface display of a co-factor containing enzyme,ω-transaminase fromVibriofluvialisusing theBacillussubtilisspore display system[J].Biosci Biotech Biochem,2011,75(9):1862-1865.
[17] MIDELFORT K S,KUMAR R,HAN S,et al.Redesigning and characterizing the substrate specificity and activity ofVibriofluvialisaminotransferase for the synthesis of imagabalin[J].Protein Eng Des Sel,2013,26(1):25-33.
[18] YUN H,LIM S,CHO B K,et al.ω-Amino acid:pyruvate transaminase fromAlcaligenesdenitrificansY2k-2:a new catalyst for kinetic resolution ofβ-amino acids and amines[J].Appl Environ Microbiol,2004,70(6):2529-2534.
[19] GREEN A P,TURNER N J,O'REILLY E.Chiral amine synthesis usingω-transaminases:an amine donor that displaces equilibria and enables high-throughput screening[J].Angew Chem Int Ed,2014,53(40):10714-10717.
[20] MALIK M S,PARK E S,SHIN J S.ω-Transaminase-catalyzed kinetic resolution of chiral amines using L-threonine as an amino acceptor precursor[J].Green Chem,2012,14(8):2137-2140.
[21] STEFFEN-MUNSBERG F,MATZEL P,SOWA M A,et al.Bacillusanthracisω-amino acid:pyruvate transaminase employs a different mechanism for dual substrate recognition than other amine transaminases[J].Appl Microbiol Biotechnol,2016,100(10):4511-4521.
[22] HOPWOOD J,TRUPPO M D,TURNER N J,et al.A fast and sensitive assay for measuring theactivity and enantioselectivity of transaminases[J].Chem Commun,2011,47(2):773-775.
[24] MALIK M S,PARK E S,SHIN J S.Features and technical applications ofω-transaminases[J].Appl Microbiol Biotechnol,2012,94(5):1163-1171.
[25] HWANG B Y,KIM B G.High-throughput screening method for the identification of active and enantioselectiveω-transaminases[J].Enzyme Microb Technol,2004,34(5):429-436.
[26] MATHEW S,SHIN G,SHON M,et al.High throughput screening methods forω-transaminases[J].Biotechnol Bioproc Eng,2013,18(1):1-7.
[27] SPAHN H,LANGGUTH P.Chiral amines derived from 2-arylpropionic acids-novel reagents for the liquid-chromatographic (LC) fluorescence assay of optically-active carboxylic-acid xenobiotics[J].Pharm Res,1990,7(12):1262-1268.
[28] BEA H S,LEE S H,YUN H.Asymmetric synthesis of (R)-3-fluoroalanine from 3-fluoropyruvate usingω-transaminase[J].Biotechnol Bioproc Eng,2011,16(2):291-296.
[29] JANES L E,KAZLAUSKAS R J,QUICK E.A fast spectrophotometric method to measure the enantioselectivity of hydrolases[J].J Org Chem,1997,62(14):4560-4561.
[30] WEISS M S,PAVLIDIS I V,VICKERS C,et al.Glycine oxidase based high-throughput solid-phase assay for substrate profiling and directed evolution of (R)- and (S)-selective amine transaminases[J].Anal Chem,2014,86(23):11847-11853.
[31] MIRIYALA B,BHATTACHARYYA S,WILLIAMSON J S.Chemoselective reductive alkylation of ammonia with carbonyl compounds:synthesis of primary and symmetrical secondary amines[J].Tetrahedron,2004,60(7):1463-1471.
[32] BAUD D,LADKAU N,MOODY T S,et al.A rapid,sensitive colorimetric assay for the high-throughput screening of transaminases in liquid or solid-phase[J].Chem Commun,2015,51(97):17225-17228.
[34] SCHEIDT T,LAND H,ANDERSON M,et al.Fluorescence-based kinetic assay for high-throughput discovery and engineering of stereoselectiveω-transaminases[J].Adv Synth Catal,2015,357(8):1721-1731.
[35] REHN G,AYRES B,ADLERCREUTZ P,et al.An improved process for biocatalytic asymmetric amine synthesis byinsituproduct removal using a supported liquid membrane[J].J Mol Catal B:Enzymatic,2016,123:1-7.
[36] REHN G,ADLERCREUTZ P,GREY C.Supported liquid membrane as a novel tool for driving the equilibrium ofω-transaminase catalyzed asymmetric synthesis[J].J Biotechnol,2014,179(1):50-55.
[37] TRUPPO M D,ROZZELL J D,MOORE J C,et al.Rapid screening and scale-up of transaminase catalyzed reactions[J].Org Biomol Chem,2009,7(2):395-398.
[38] WU X,FEI M,CHEN Y,et al.Enzymatic synthesis of L-norephedrine by coupling recombinant pyruvate decarboxylase andω-transaminase[J].Appl Microbiol Biotechnol,2014,98(17):7399-7408.
[39] KROUTIL W,FISCHEREDER E M,FUCHS C S,et al.Asymmetric preparation of prim-,sec-,andtert-amines employing selected biocatalysts[J].Org Pro Res Dev,2013,13(5):751-759.
[40] SIMON R C,RICHTER,N,BUSTO E,et al.Recent developments of cascade reactions involvingω-transaminases[J].ACS Catal,2014,4(1):129-143.
[41] KOSZELEWSKI D,CLAY D,ROZZELL D,et al.Deracemisation of alpha-chiral primary amines by a one-pot,two-step cascade reaction catalyzed byω-transaminases[J].Eur J Org Chem,2009(14):2289-2292.
[42] FRANCE S P,HUSSAIN S,HILL A M,et al.One-pot cascade synthesis of mono- and disubstituted piperidines and pyrrolidines using carboxylic acid reductase (CAR),ω-transaminase (ω-TA),and imine reductase (IRED) biocatalysts[J].ACS Catal,2016,6(6):3753-3759.
[43] SHIN G,MATHEW S,SHON M,et al.One-pot one-step deracemization of amines usingω-transaminases[J].Chem Commun,2013,49(77):8629-8631.
[44] SHIN J S,KIM B G.Transaminase-catalyzed asymmetric synthesis of L-2-aminobutyric acid from achiral reactants[J].Biotechnol Lett,2009,31(10):1595-1599.
[45] PARK E,KIM M,SHIN J S.One-pot conversion of L-threonine into L-homoalanine:biocatalytic production of an unnatural amino acid from a natural one[J].Adv Synth Catal,2010,352(18):3391-3398.
[46] BEA H S,SEO Y M,CHA M H,et al.Kinetic resolution of alpha-methylbenzylamine by recombinantPichiapastorisexpressingω-transaminase[J].Biotechnol Bioproc Eng,2010,15(3):429-434.
[47] SEO Y M,MATHEW S,BEA H S,et al.Deracemization of unnatural amino acid:homoalanine using D-amino acid oxidase andω-transaminase[J].Org Biomol Chem,2012,10(12):2482-2485.
[48] CHO B K,CHO H J,PARK S H,et al.Simultaneous synthesis of enantiomerically pure (S)-amino acids and (R)-amines using coupled transaminase reactions[J].Biotechnol Bioeng,2003,81(7):783-789.
[49] KIM J,KYUNG D,YUN H,etal.Cloning and characterization of a novelβ-transaminase fromMesorhizobiumsp strain LUK:a new biocatalyst for the synthesis of enantiomerically pureβ-amino acids[J].Appl Environ Microbiol,2007,73(6):1772-1782.
[50] BEA H S,PARK H J,LEE S H,et al.Kinetic resolution of aromatic beta-amino acids byω-transaminase[J].Chem Commun,2011,47(20):5894-5896.
