氧化吲哚螺环作为多种疾病治疗药物的研究前景
2018-06-05金勇哲师健友
金勇哲,魏 鑫,师健友△
(1.四川省医学科学院·四川省人民医院,个体化药物治疗四川省重点实验室,四川 成都 610072;2.电子科技大学医学院,四川 成都 610054)
氧化吲哚螺环是药物化学,天然药物化学,有机化学等多个领域被广泛研究药物母核结构(见图1)。近年来关于该结构的相关研究,特别是药理学、生物活性、有机合成的研究一直都是热点领域。该分子结构可以形成一个稳定的三环体系,这些化合物在2-位氧化吲哚支架的基础上,通过3-位螺环形成非平面的三环或多环骨架,所构成的饱和或非饱和杂环化合物是许多药物和天然产物的核心结构[1]。一方面以氧化吲哚螺环为核心结构的许多具有药理活性的生物碱已经被发现,如horsfiline,mitraphylline,spirotryprotatins A、B(见图2)等。另一方面,人工设计合成的氧化吲哚螺环小分子由于非平面,特别是螺杂环结构具有的高蛋白质亲和力,使其可以作为生物靶向药物,同样具有非常广泛的药物研发前景。这类结构主要报道的药理活性有:抗肿瘤活性,抗炎及解热镇痛活性,抗病毒活性,抗结核活性,抗真菌活性,治疗贫血的药理活性等[2~5]。其中,抗肿瘤活性的研究最为深入。本文重点介绍近年来氧化吲哚螺环结构的相关先导化合物及药物药理活性研究进展,并对该类结构先导化合物可作为多种疾病治疗药物的研究前景作以展望。

图1 氧化吲哚螺环结构
1 氧化吲哚螺环化合物的药理活性研究进展
1.1抗肿瘤活性癌症是全球第二大死亡原因,2015年有880万人死于癌症。全球范围内,近1/6的死亡是由癌症引起的,新病例的数量预计在未来20年内将增加约70%。癌变是一种是由许多致癌物质导致的,高度复杂的多步骤诱导过程。在癌症中,细胞分裂和生长失控,形成恶性肿瘤,可能侵袭附近的正常细胞,甚至通过淋巴扩散到身体较远部位的系统或血液[6]。
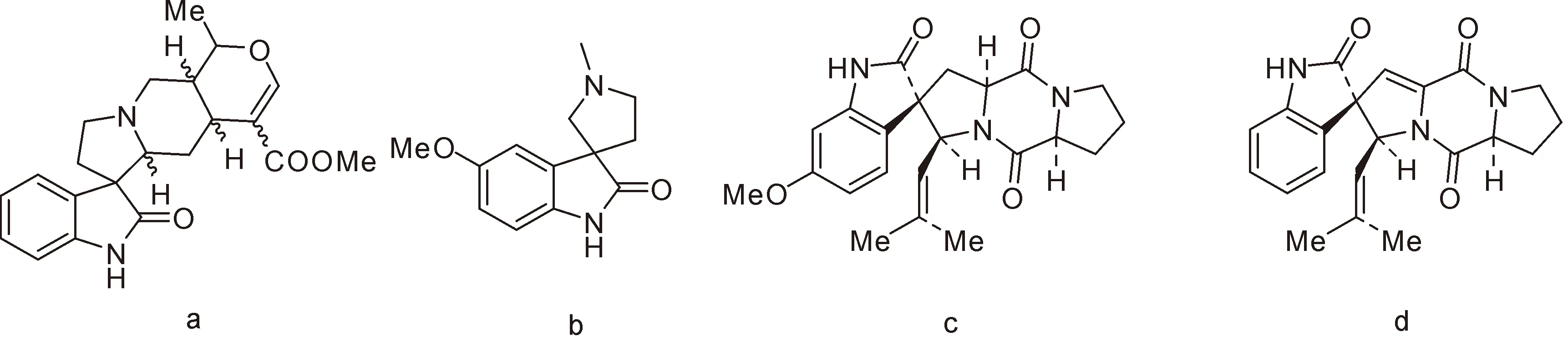
图2 具有药理活性的氧化吲哚螺环生物碱结构 (a.mitraphylline;b.horsfiline;c.spirotryprotatins A;d.spirotryprotatins B)
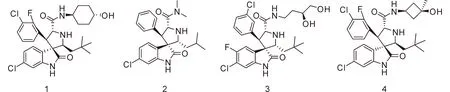
如图3所示,通过阻断p53蛋白MDM2蛋白相互作用,设计一种MDM2蛋白小分子抑制剂,已成为一种有效的癌症治疗策略。近年来,高效、高选择性、有效的MDM2表达抑制剂已成功获得,并已有化合物进入癌症治疗的临床试验阶段[7]。如图4所示,是小分子MDM2抑制剂1与MDM2的共晶结构,该化合物由密歇根大学Wang Shaomeng课题组发现,并在2012年由赛诺菲公司进行了临床试验。该化合物1是从2005年首次报道的化合物2经过多次优化得到,具有氧化吲哚螺环结构。从2首先优化得到的先导化合物3与MDM2结合较好(Ki = 23.5 nM)并展现了良好的肿瘤抑制活性(IC50≈1 μM),化合物4(MI - 888)的氧化吲哚螺环支架与3有不同的立体化学结构,且化合物4在接种肿瘤大鼠模型上表现出了良好的抗肿瘤效果,且无毒性迹象。化合物4进一步基团优化最终得到化合物1。

图3 MDM2和MDMX对p53的抑制作用[8]

图4 化合物1和MDM2的共晶结构 [8]
PLK4是另一个近年来被广泛研究的抗肿瘤药物靶点,它是一种独特的丝氨酸激酶家族的成员,是中心粒复制的主要调节因子,对保持基因组完整性至关重要,已在多种人类肿瘤的发生中观察到PLK4的过表达。小分子化合物5[8],作为第一个有效PLK4抑制剂在2013年被Sampson等发现,其在各种临床前肿瘤模型中显示出显著的抗肿瘤活性。该先导药物已由加拿大卫生部和美国食品药品管理局批准进入人实体瘤的临床试验治疗阶段。在此基础上,Sampson等在2015年基于PLK4晶体结构的模型计算设计并合成了一系列化合物,不仅有很好的PLK4亲和力和抗增殖活性,且具有较好的物理化学特性和药物动力学性质[9]。
Yu等[10]在2014年设计了两类新型甾体螺环吡咯烷基氧化吲哚化合物,从脱氢表雄酮利用1,3-偶极环加成反应完成了化合物合成,并进一步评估他们对四种人类肿瘤细胞(MGC-803、EC109、SMMC-7721和 MCF-7)的抗增殖活性。生物评价结果表明,这些合成的甾体类螺吡咯烷基羟吲哚化合物具有良好抗肿瘤活性。特别是,化合物6对人肝癌细胞表现出良好的抗增殖活性(IC50= 0.71 μM)。Bin等[11]也发现系列化合物中的4f和4I的在对肿瘤细胞T24与MGC-803的抑制效果上比阳性对照药物5-FU更有效,其IC50值分别为4.43 μM和8.45 μM。

Dong等[12]在2015年通过细胞系的细胞毒活性化合物的筛选发现系列化合物,特别是化合物7具有良好的抗肿瘤作用和诱导凋亡作用。且该研究提供了一种有效快速建立具有恶唑酮结构的螺环吲哚衍生物化合物库的方法,为先导化合物的筛选提供了更多选择。
Kumar等[13,14]在2015年根据生物电子等排体原则,合成设计(MI-63/219)的系列类似物8(见图8),是一种能与p53相互作用的MDM2小分子抑制剂,报道中的化合物在MCF-7裸鼠移植瘤的抗增殖试验中表现出很好的活性,是一种有很大发展潜力的抗乳腺癌药物。
Monteiro等[15]由1,3-偶极环加成反应合成了一系列新型的氧化吲哚螺环衍生物9(见图8)。化合物筛选的体外细胞活性试验表明对MCF-7乳腺癌细胞系有抑制增殖作用。这十九个化合物的活性实验中,六种GI50低于12 μM。其中两个化合物对MCF-7肿瘤细胞和MDA-MB-231肿瘤细胞有高度选择性。更重要的是,它们在非肿瘤细胞系HEK 293T的毒性实验中没有表现出明显毒性。

Hamblett等[16]在2007年设计一种羟肟酸衍生物类螺环化合物10,其具有取代氧化吲哚螺环母核结构,可作为组蛋白去乙酰化酶(HDAC)抑制剂,抑制组蛋白去乙酰化酶可以选择性地诱导终末分化以及抑制赘生性细胞的增殖,用于癌症的治疗。因此,该化合物可用于治疗赘生性细胞增殖的肿瘤患者,以及预防和治疗硫氧化蛋白(Trx)介导的疾病,如自身免疫性疾病、变应性疾病和炎性疾病,还可用于预防或治疗中枢神经系统(CNS)疾病,例如神经变性疾病。
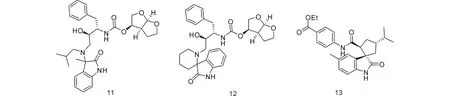
1.2抗病毒药理活性人体免疫缺陷病毒(HIV)是一种能感染免疫细胞的逆转录病毒,它会破坏人体免疫系统或损害其功能。HIV感染导致的获得性免疫缺陷综合征(AIDS),是一种威胁全球健康的疾病。据统计,2016年全球HIV感染患者约有3670万人,仅2016年全年就有约100万人死于HIV感染[19]。目前在FDA批准的药物中,氧化吲哚结构可以有效地相互作用并填充P2’ HIV蛋白酶活性区。Ghosh等[17]据此设计并合成了新型氧吲哚衍生物。这项工作产生了FDA批准应用于临床治疗的第二代蛋白酶抑制剂。11已经显示出抗病毒,其两种非对映体的Ki仅有微小的差异,分别为6 nM和3 nM。考虑到HIV蛋白酶活性位点的约束明显改善了酶抑制活性,12作为P2’-配体的螺环氧吲哚衍生物的可行性被实验验证。然而,其抑制效果显著降低[18~20]。
此外,病毒蛋白R (Vpr)是一种编码在中央区域的小蛋白质,它也是抑制HIV-1复制的理想靶点[21]。Aida等[21,22]发展了新的方法来筛选先导化合物。从转染的COS-7细胞中纯化重组Vpr的化合物。在这些化合物中,化合物13(spirooxindole SIP-1)明显抑制Vpr功能。通过对其功能域的结合,从而显示出对HIV的高抑制活性,其IC50=0.5 μM。此外,13没有细胞毒性作用,也不破坏细胞周期。并且通过MTT测量,可以对Molt-4和HeLa细胞系也有诱导凋亡作用。
登革热是由蚊子传播的最快的病毒性疾病,全球大约有一半的人会受到威胁,全世界每年约有近4亿人感染。目前有几种有希望的疫苗用于临床,如赛诺菲公司开发的第一个疫苗Dengvaxia[23]。为了开发安全有效的抗病毒治疗药物,Wang等[24]利用了诺华化合物库的高通量表型筛选,确定了氧化吲哚螺环可作为一种新型的抗登革热药物化学结构。发现并合成了先导化合物14。实验显示该化合物抑制登革热病毒血清DENV-2的复制,EC50值为14 nM。因为14是外消旋体,对映体随后由手性柱HPLC分离,得到的 (R)-14,其EC50值为12 nM,比Senantiomer强83倍。进一步的抗病毒谱分析(R)-14对抗一组病毒,令人惊讶的是,(R)-14只抑制了DENV-2和3,但没有抑制DENV-1和4等其他病毒血清型,抗性分析和直接配体蛋白结合实验证实了这种变化。

丙型肝炎病毒(HCV)感染了超过3%的世界人口,它会导致脂肪变性、肝硬化和肝细胞癌的风险增加。丙肝病毒非结构蛋白5A (NS5A)是一种具有锌结合和脯氨酸的亲水磷蛋白。这在病毒复制周期中起着关键作用,促进了基因组的复制[25]。Zhang等[26]在2013年,以此为靶点的新型的NS5A系列抑制剂,如15和16,对HCV1b具有良好的EC50值,分别为4.95nM和0.45 nM。
1.3抗炎活性螺环吲哚衍生物化合物也可用作为非甾体抗炎药,其代表化合物17具有消炎、解热及镇痛作用,特别是对慢性疼痛有良好的治疗效果[27]。Sun等[28在研究中发现,化合物18(jp-8 g)在小鼠模型有有效体内抗炎活性。进一步的研究表明,化合物18可能是通过影响一氧化氮合酶信号通路从而产生抗炎活性。他们的研究结果表明,这些吡喃并嘧啶类化合物吲哚不仅具有潜在的癌症治疗也具有炎症治疗的潜力。

绒毛钩藤在南美被广泛应用于治疗许多与炎症相关的疾病。Rojas-Duran等[29]在2012年发现,氧化吲哚类生物碱化合物19(Mitraphylline)是这种植物的树皮主要氯仿提取物,通过小鼠模型体内的炎症过程进行了活性测试,表现出了抗炎活性。Mitraphylline被认为是一个新的可用于抗炎治疗的先导化合物。

Eastwood等[30]在2011年根据分子模拟对接设计并合成了一类化合物,如图5所示,其中20是一个强有力的和高选择性的p38α抑制剂。在激发状态,p38α磷酸化了一系列的胞内蛋白质基质,转录后调节细胞因子TNFα和IL-1B的生物合成。TNFα和IL-1B的生成在炎症疾病的发展过程中被认为是有重要影响的。因此,该化合物高选择性的抑制p38α使其有望发展为一种新型抗炎剂用于临床治疗。

图5 p38α抑制剂和p38α晶体结构的氢键作用的分子结构式
1.4抗真菌活性Haddad等[35]在2015年报道了一系列的氧化吲哚螺环化合物21。这些新型杂环化合物的立体化学结构已经被单晶衍射研究证实。化合物在体外的抗菌作用进行了试验筛选,表现出了一定抗真菌、抗疟和抗结核活性。其中的几种化合物活性与阳性标准药物相当。
Wu等[32]在2015年报道了通过氧化吲哚衍生物和芳香丙烯腈衍生物的双Michael反应,在温和的条件下生成系列氧化吲哚四氢呋喃螺环衍生物。随后,对所有合成的系列化合物的抗真菌活性进行了评价。初步结果表明该系列结构有一定的抗真菌活性,且化合物22的抗真菌活性最好,和阳性对照药物相近。
1.5抗结核活性2016年Rouatbi等[33]报道了从靛红衍生物和L-脯氨酸原位生成非稳定的叶立德,与1,3-偶极子环加成反应生成一系列新型功能化双螺环化合物。该类化合物对结核分枝杆菌的体外筛选试验显示出良好的活性。其中具有氧化吲哚螺环结构的化合物23被认为是很有发展潜力的抗结核药物。
1.6治疗贫血药理活性Fletcher等[34]于2008年报道了一系列氮杂吲哚螺环化合物24,作为缺氧诱导因子(HIF)羟化酶抑制剂用于抑制低氧诱导因子羟化酶活性,特别在治疗与癌症相关的贫血有效,但对红细胞生成素不具治疗作用。Fletcher等[35]在2009年优化了此类螺环化合物,通过在不同的动物癌症贫血模型中增加促红细胞生成素(EPO)水平、红细胞计数、血红蛋白水平和血细胞比容。进一步证实此类化合物对治疗和预防癌症贫血或与癌症相关的贫血的发展有效。
Vachal等[36]在2012年发现,初始苗头化合物中,一系列螺环吲哚衍生物25作为短效PHDi抑制剂,通过抑制脯氨酰羟化酶并得到理想的的PK/PD数据,证明有一定的治疗贫血疗效。并以该类化合物为基础,进一步结构优化得到了治疗效果更好的先导化合物。
2.7抗疟疾药理活性先导化合物26(NITD609)是Rottmann等[37]在2010年合成的能够替代青蒿素类的抗疟药物,对抗恶性疟原虫临床分离株时表现了很好的药物活性,其IC50在0.5~1.4 nM,该先导药物能够迅速降低恶性疟原虫的蛋白合成。具有良好的物理化学性质,在一定剂量下具有良好的安全性,药代动力学特性和药理活性。该先导化合物最近已完成了二期临床试验。(NCT01860989;NCT01836458;NCT01524341)。

2 前景与展望
综上所述,由于氧化吲哚螺环结构在抗肿瘤,抗炎及解热镇痛,抗病毒,抗结核,抗真菌,抗疟疾,治疗贫血等多个方面表现出良好的药理活性,使其具有极好的医药研究前景,可作为多种疾病的治疗药物进一步开展研究。近年来,随着更多更深入的活性研究不断被文献报道,特别是抗肿瘤靶向小分子药物领域,拥有氧化吲哚螺环结构的靶向先导化合物不断被发现,由于其不仅具有较好的靶向选择性,且拥有良好的理化性质及药代动力学性质,已有多个先导药物进入临床研究阶段。在AIDS,炎症,疟疾等多种疾病的治疗中,均有氧化吲哚螺环结构的先导化合物进入临床研究。另一方面,由于医药领域的研究需求,关于该类结构有机合成方法的研究也在不断深入,丰富、高效、快速的合成方法也为这类结构的发现和合成研究提供很大的便利。可以预见,在以后,在多种疾病的治疗药物研发领域都可以看到氧化吲哚螺环结构的相关报道,该类结构的药理活性研究及其进一步的临床药理研究仍将是一个热点研究领域。
[1] Brown RT.Indoles,the monoterpenoid indole alkaloids[J].The Chemistry of Heterocyclic Compounds,1983,25(Part 4):85.
[2] Yu B,Yu DQ,Liu HM.Spirooxindoles:Promising scaffolds for anticancer agents[J].European journal of medicinal chemistry,2015,97:673-698.
[3] Santos MMM.Recent advances in the synthesis of biologically active spirooxindoles[J].Tetrahedron,2014,52(70):9735-9757.
[4] Pavlovska TL,Redkin RG,Lipson VV,et al.Molecular diversity of spirooxindoles.Synthesis and biological activity[J].Molecular diversity,2016,20(1):299-344.
[5] Ye N,Chen H,Wold EA,et al.Therapeutic potential of spirooxindoles as antiviral agents[J].ACS infectious diseases,2016,2(6):382-392.
[6] Irigaray P,Belpomme D.Basic properties and molecular mechanisms of exogenous chemical carcinogens[J].Carcinogenesis,2009,31(2):135-148.
[7] Zhao Y,Aguilar A,Bernard D,et al.Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment:miniperspective[J].Journal of medicinal chemistry,2014,58(3):1038-1052.
[8] Mason J M,Lin D C C,Wei X,et al.Functional characterization of CFI-400945,a Polo-like kinase 4 inhibitor,as a potential anticancer agent[J].Cancer cell,2014,26(2):163-176.
[9]Sampson PB,Liu Y,Patel NK,et al.The Discovery of Polo-Like Kinase 4 Inhibitors:Design and Optimization of Spiro [cyclopropane-1,3’[3 H] indol]-2’(1’H)-ones as Orally Bioavailable Antitumor Agents[J].Journal of medicinal chemistry,2014,58(1):130-146.
[10]Yu B,Shi XJ,Qi PP,et al.Design,synthesis and biological evaluation of novel steroidal spiro-oxindoles as potent antiproliferative agents[J].The Journal of steroid biochemistry and molecular biology,2014,141:121-134.
[11]Yu B,Qi PP,Shi XJ,et al.Discovery of novel steroidal pyran-oxindole hybrids as cytotoxic agents[J].Steroids,2014,88:44-52.
[12]Dong H,Song S,Li J,et al.The discovery of oxazolones-grafted spirooxindoles via three-component diversity oriented synthesis and their preliminary biological evaluation[J].Bioorganic & medicinal chemistry letters,2015,25(17):3585-3591.
[13]Siegel RL,Miller KD,Jemal A.Cancer statistics,2015[J].CA:a cancer journal for clinicians,2015,65(1):5-29.
[14]Kumar A,Gupta G,Bishnoi AK,et al.Design and synthesis of new bioisosteres of spirooxindoles (MI-63/219) as anti-breast cancer agents[J].Bioorganic & medicinal chemistry,2015,23(4):839-848.
[16]Hamblett C,kattar S,Mampreian D,et al.Aryl-Fused Spirocyclic compounds[P].WO2007136605,2007-04-14.
[17] Ghosh AK,Schiltz G,Perali RS,et al.Design and Synthesis of Novel HIV-1 Protease Inhibitors Incorporating Oxyindoles as the P2’-Ligands[J].Bioorganic & Medicinal Chemistry Letters,2006,16(7):1869-1873.
[18] Ghosh AK,Swanson LM,Cho H,et al.Structure-based design:synthesis and biological evaluation of a series of novel cycloamide-derived HIV-1 protease inhibitors[J].Journal of medicinal chemistry,2005,48(10):3576-3585.
[19] Reid RC,Pattenden LK,Tyndall JDA,et al.Countering cooperative effects in protease inhibitors using constrained β-strand-mimicking templates in focused combinatorial libraries[J].Journal of medicinal chemistry,2004,47(7):1641-1651.
[20] Glenn MP,Pattenden LK,Reid RC,et al.β-Strand mimicking macrocyclic amino acids:templates for protease inhibitors with antiviral activity[J].Journal of medicinal chemistry,2002,45(2):371-381.
[21] Aida Y,Matsuda G.Role of Vpr in HIV-1 nuclear import:therapeutic implications[J].Current HIV research,2009,7(2):136-143.
[22] Hagiwara K,Murakami T,Xue G,et al.Identification of a novel Vpr-binding compound that inhibits HIV-1 multiplication in macrophages by chemical array[J].Biochemical and biophysical research communications,2010,403(1):40-45.
[23] Fink K,Shi PY.Live attenuated vaccine:the first clinically approved dengue vaccine[J].Expert Review of Vaccines,2014,13 (2):185-188.
[24] Wang Q Y,Dong H,Zou B,et al.Discovery of dengue virus NS4B inhibitors[J].Journal of virology,2015,89(16):8233-8244.
[25] Belda O,Targett-Adams P.Small molecule inhibitors of the hepatitis C virus-encoded NS5A protein[J].Virus research,2012,170(1-2):1-14.
[26] Zhang Y,Zhang J,Xie H,et al.Pyrrolidine derivative used as hepatitis c inhibitor and application thereof inmedicine[P].2013,CN 103420991
[27]BergeO.G.,Claesson A.,Swahn B.M.,et al.New compounds[P].WO2001005790,2001-01-25
[28]Sun Y,Liu J,Sun T,et al.Anti-cancer small molecule JP-8g exhibits potent in vivo anti-inflammatory activity[J].Scientific reports,2014,4:4372.
[29] Rojas-Duran R,González-Aspajo G,Ruiz-Martel C,et al.Anti-inflammatory activity of Mitraphylline isolated from Uncaria tomentosa bark[J].Journal of ethnopharmacology,2012,143(3):801-804.
[30]Eastwood P,González J,Gómez E,et al.Indolin-2-one p38α inhibitors I:Design,profiling and crystallographic binding mode[J].Bioorganic & medicinal chemistry letters,2011,21(14):4130-4133.
[31]Haddad S,Boudriga S,Akhaja TN,et al.A strategic approach to the synthesis of functionalized spirooxindole pyrrolidine derivatives:in vitro antibacterial,antifungal,antimalarial and antitubercular studies[J].New Journal of Chemistry,2015,39(1):520-528.
[32]Wu JS,Zhang X,Zhang YL,et al.Synthesis and antifungal activities of novel polyheterocyclic spirooxindole derivatives[J].Organic & biomolecular chemistry,2015,13(17):4967-4975.
[33]Rouatbi F,Askri M,Nana F,et al.Synthesis of new spirooxindole derivatives through 1,3-dipolar cycloaddition of azomethine ylides and their antitubercular activity[J].Tetrahedron Letters,2016,57(2):163-167.
[34]Fletcher JM,Hale JJ,Miao S,et al.Spiroindalones[P].WO2008144266.2008-11-27.
[35]Fletcher JS,Hale JJ,Miao S,et al.Spiroanaindoles[P].WO2009137291.2009-11-12.
[36]Vachal P,Miao S,Pierce JM,et al.1,3,8-Triazaspiro [4.5] decane-2,4-diones as efficacious pan-inhibitors of hypoxia-inducible factor prolyl hydroxylase 1-3 (HIF PHD1-3) for the treatment of anemia[J].Journal of medicinal chemistry,2012,55(7):2945-2959.
[37] Rottmann M,McNamara C,Yeung BKS,et al.Spiroindolones,a potent compound class for the treatment of malaria[J].Science,2010,329(5996):1175-1180.
