儿童Bardet-Biedl综合征1例病例报告
2018-05-28丁静超王春林方燕兰
丁静超 王春林 方燕兰 梁 黎
1 病例资料
男,12岁5个月。因“阴茎短小10余年,生长迟缓2年余”就诊于浙江大学医学院附属第一医院(我院)儿科。
患儿系G1P1,足月顺产,产时无窒息抢救史,出生体重3.5 kg,身长50 cm。否认母孕期疾病史及特殊药物应用史。生后在外院诊断为阴茎短小和左脚多趾畸形,1岁时行左脚小趾旁多趾畸形切除手术,阴茎短小随年龄增长未见明显改善。生后体重一直增长较快,年增长4~6 kg,较同龄儿童体型明显肥胖,外院曾诊断为肥胖,近年来经饮食控制、加强运动锻炼后体重增加有所减缓。近2年来生长缓慢,患儿家长诉身高年增长速度约每年3.5 cm。2~3岁时表现出智力低下,语言发育缓慢,计算能力差,但未行相应智能发育评估。7岁出现视物不清,逐渐加重,外院眼底检查发现视神经萎缩,未予特殊处理。患儿父母非近亲婚配,否认家族类似疾病及其他遗传代谢性疾病史。
体格检查:身高152 cm(-0.44 SD),体重50 kg,BMI 21.6 kg·m-2,肥胖体态,发音含糊,表达能力差,心、肺、腹部及神经系统查体未见异常。阴茎隐匿,长约1 cm,G1期,阴毛PH1期,手指短小,左脚外侧可见长1 cm左右的瘢痕。
辅助检查:黄体生成素释放激素(LHRH)激发试验: LH峰值2.4 mIU·mL-1,FSH峰值12.7 mIU·mL-1。睾丸B超提示,睾丸未发育(左1.6 cm×0.8 cm,右1.8 cm×1.0 cm)。眼底检查示,视网膜豹纹状。血常规、肝肾功能、甲状腺功能、皮质醇、糖化血红蛋白、血气分析、血乳酸指标均在正常值范围。
患儿色素性视网膜炎、肥胖、多趾畸形、性腺发育不全、智力低下等,符合巴德-毕氏综合征(BBS)临床诊断标准[1]。
征得患儿父母知情同意后,抽取患儿及其父母静脉血行BBS相关基因panel高通量测序以及对Sanger 测序验证。图1显示,患儿BBS2基因存在c.646C>T(p.R216X)和c.1206dupA(p.R403fs) 2个复合杂合突变位点;c.646C>T(p.R216X)来自于父亲,已被报道,突变后的氨基酸序列终止密码由第722位提前至216位;c.1206dupA(p.R403fs)来自于母亲。
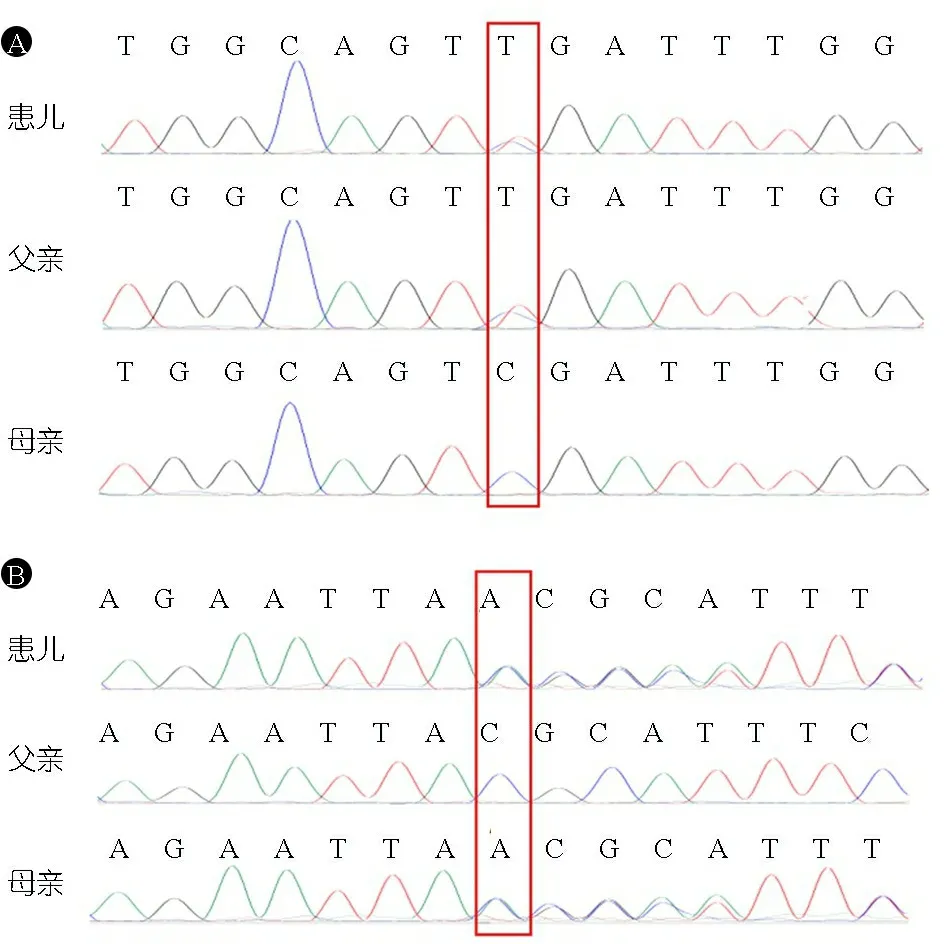
图1 患儿BBS相关基因Panel检测Sanger测序验证
注 A: BBS2 chr16-56540103 c.646C>T (p.R216X) 无义突变; B: BBS2 chr16 56535283- 56535284 c.1206dupA (p.R403fs) 移码突变
2 讨论
BBS又称性幼稚-肥胖-多趾综合征,是一组以色素性视网膜炎、肥胖、多指/趾畸形、性腺发育不全、智力低下和肾功能异常六大主要临床症状为主,可伴有肝纤维化、发育迟缓、聋哑症、矮小、内分泌紊乱以及心血管异常等次要症状的常染色体隐性遗传病。BBS的患病率在不同地区、不同民族间存在较大差异,北欧人群患病率约1∶160 000[2-4],科威特贝都因人群约1∶13500,纽芬兰约1∶175 000,我国的患病率目前尚无明确资料。本文患儿12岁5个月,明确诊断为BBS,基因测序显示c.646C>T(p.R216X)和c.1206dupA(p.R403fs),在国外文献中已有报道[5,6],在国内尚属未见报道。
目前已报道BBS相关的突变基因有21个[7, 8]。Khan等[9]在2015年对既往报道的BBS1~19的基因突变类型及表型做了总结。其后,Kinga等[10]和Schaefer等[11]相继报告了BBS20(FT172),Arif等[12]和Elise等[7]报告了BBS21(C8orF37)。21个BBS基因中,BBS1、2、6、10和12报道较多,而BBS11、15、18和19则鲜有报道,BBS20和BBS21为近年报道。在BBS基因突变类型中,以错义/无义、剪接及缺失插入突变多见,复杂重排相对发生少。目前国内报道病例大多为临床诊断,基因诊断明确的病例及家系报道均较少,故各基因型分布情况尚未可知[13],陈瑶等[14]报告1例13岁男患儿为BBS2基因纯合突变(c.1148_1149 dupTC,p.His 384 Serfs* 34)。另外,尚有2个家系报道了BBS突变,分别位于BBS5[15]和BBS7/BBS12[16]。
BBS的临床表型较多,色素性视网膜炎、肥胖、性腺发育不全、多指(趾)畸形、智力发育迟缓和肾异常为主要表现,除此之外,还有多种次要临床表现。本文在Khan等综述的基础上进一步总结了BBS基因突变患儿的表型,见表1。6种主要表现几乎可见于目前所有已报道的BBS基因型,除BBS15,因自2010年Kim 等[17]报告了BBS15后,尚未见其他关于该基因型及其临床表型的报道,而Kim等在文献中并未详细描述BBS患儿的具体临床特征。BBS的主要症状中,视网膜变性报道最多,起病早,病情进展迅速,部分患者20岁之前就已完全失明[18, 19]。肥胖症在BBS患儿中亦比较常见,机制可能与BBS蛋白参与能量平衡调节相关。最新的一项小鼠动物实验中证实,BBS1基因的选择性表达导致能量代谢失衡,摄入的增加及能量消耗的减少最终导致小鼠发生肥胖[20]。性腺的发育异常在BBS男女中均可见,在男性中多表现为小阴茎及小睾丸,在女性既可表现为输卵管、子宫和卵巢等发育不全,亦可表现为泌尿生殖窦、膀胱阴道瘘及尿道口缺如等[21]。多指/趾畸形是本病早期即可观察到的临床表型,表现形式多样,可累及四肢,也可表现在单一肢体。其机制可能与Sonic Hedgehog(SHH)信号通路的损害有关,而这一通路在肢体发育及机体左右对称性发育方面具有重要作用[22]。一半以上的BBS患儿存在智力发育迟缓,可表现为语言、智力、行为等多方面发育水平低于同龄儿童。其机制可能与初级纤毛损害,进而影响神经信号发生、传导以及海马发育等有关[21]。Brinckman等[23]在对42例2~61岁的BBS患者进行神经心理测验,发现大多数受试者存在智力缺陷和精细运动受损等问题。肾脏异常在BBS患儿中发生率相对较低,多数并无解剖结构异常,仅表现为尿液浓缩功能受损[24]。其机制为纤毛功能受损导致肾脏小管水通道异常,水重吸收功能障碍,最终导致尿液浓缩功能受损[25]。BBS的次要症状众多,但各类型间发生率相差较大。表1显示,肝脏纤维化、面部畸形、发育迟缓、心脏疾病、嗅觉异常及耳聋等临床表现在较多的基因型中均被报道;糖尿病、神经运动疾病、高血压及呼吸道疾病亦可见于较多基因型,而不规则牙和甲状腺功能低下等见于较少基因型。
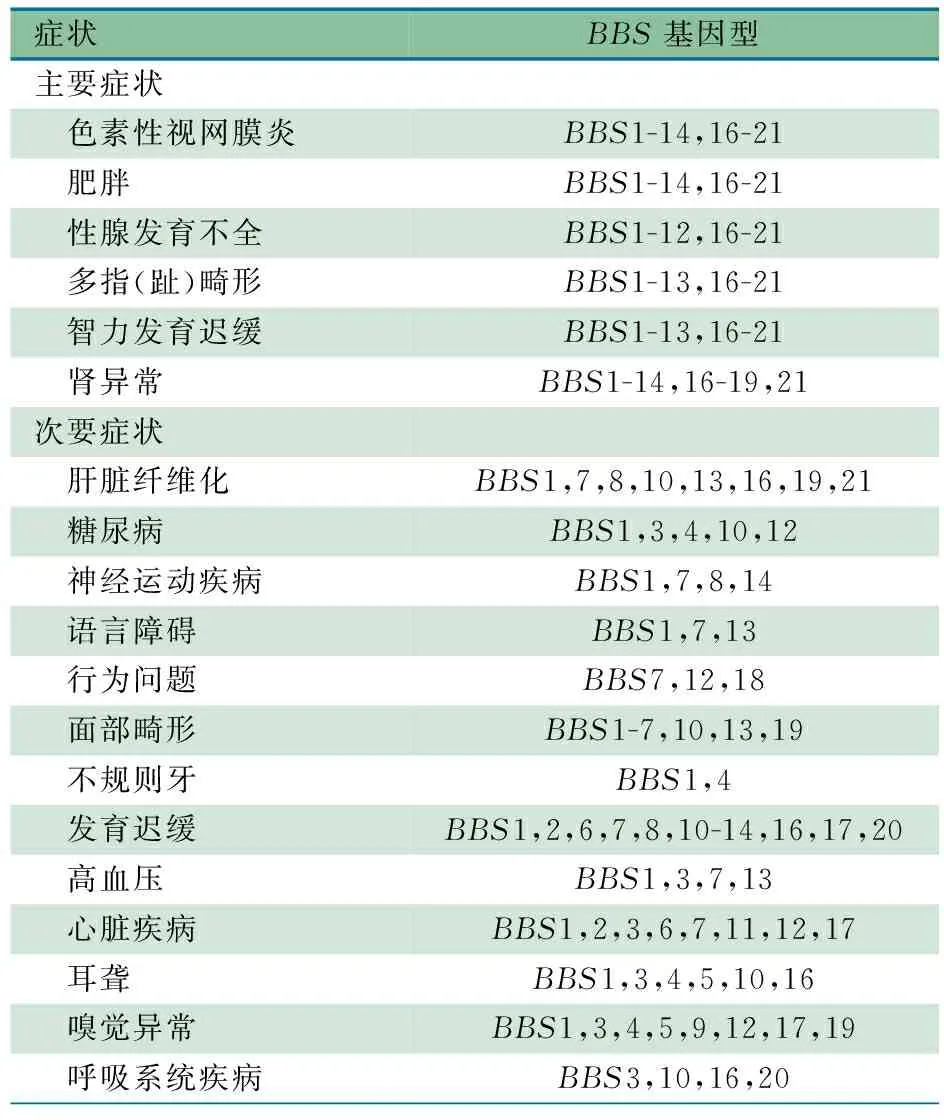
表1 不同BBS基因突变患儿的表型
对于BBS,目前临床上并无特效治疗,主要为对症治疗,早期诊断对改善患儿生活质量有一定的帮助。
参考文献
[1] Beales PL, Elcioglu N, Woolf AS, et al. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J MED Genet, 1999, 36(6): 437-446
[2] M'hamdi O, Ouertani I, Chaabouni-Bouhamed H. Update on the genetics of bardet-biedl syndrome. MOL Syndromol, 2014, 5(2): 51-56
[3] Farag TI, Teebi AS. High incidence of Bardet Biedl syndrome among the Bedouin. Clin Genet, 1989, 36(6): 463-464
[4] Moore SJ, Green JS, Fan Y, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A, 2005, 132(4): 352-360
[5] Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a mendelian recessive disorder. Science, 2001, 293(5538): 2256-2259
[6] Nishimura DY, Searby CC, Carmi R, et al. Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome(BBS2). Hum Mol Genet, 2001, 10(8): 865-874
[7] Heon E, Kim G, Qin S, et al. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum Mol Genet, 2016, 25(11): 2283-2294
[8] Priya S, Nampoothiri S, Sen P, et al. Bardet-Biedl syndrome: genetics, molecular pathophysiology, and disease management. Indian J Ophthalmol, 2016, 64(9): 620-627
[9] Khan SA, Muhammad N, Khan MA, et al. Genetics of human Bardet-Biedl syndrome, an updates. Clin Genet, 2016, 90(1): 3-15
[10] Bujakowska KM, Zhang Q, Siemiatkowska AM, et al. Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum Mol Genet, 2015, 24(1): 230-242
[11] Schaefer E, Stoetzel C, Scheidecker S, et al. Identification of a novel mutation confirms the implication of IFT172 (BBS20) in Bardet-Biedl syndrome. J Hum Genet, 2016, 61(5): 447-450
[12] Khan AO, Decker E, Bachmann N, et al. C8orf37 is mutated in Bardet-Biedl syndrome and constitutes a locus allelic to non-syndromic retinal dystrophies. Ophthalmic Genet, 2016, 37(3): 290-293
[13] 沈涛, 严新民, 肖春杰. 巴德-毕氏综合征研究的现状及意义. 中华医学遗传学杂志, 2013, 30(5):570-573
[14] 陈瑶, 李娟, 王剑, 等. Bardet-Biedl 综合征1 例报告并文献复习. 临床儿科杂志, 2017, 35(1):28-32
[15] 刘兵, 杨洋, 林婴. 一个中国Bardet-Biedl综合征与BBS5位点连锁. 现代预防医学, 2008, 35(9):1738-1741
[16] Yang Z, Yang Y, Zhao P. A novel mutation in BBS7 gene cause Bardet Biedl syndrome in a Chinese family. Mol Vis, 2008, 14(266-68): 2304-2308
[17] Kim SK, Shindo A, Park TJ, et al. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science, 2010, 329(5997): 1337-1340
[18] Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet, 2013, 21(1): 8-13
[19] Mockel A, Perdomo Y, Stutzmann F, et al. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Prog Retin Eye Res, 2011, 30(4): 258-274
[20] Guo DF, Cui H, Zhang Q, et al. The bbsome controls energy homeostasis by mediating the transport of the leptin receptor to the plasma membrane. PLoS Genet, 2016, 12(2): e1005890
[22] 张艳, 徐选福, 郭传勇. Sonic Hedgehog基因及其在发育过程中的调控作用. 现代生物医学进展, 2014, 14(2):358-360
[23] Brinckman DD, Keppler-Noreuil KM, Blumhorst C, et al. Cognitive, sensory, and psychosocial characteristics in patients with Bardet-Biedl syndrome. Am J Med Genet A, 2013, 161(12): 2964-2971
[24] Putoux A, Attie-Bitach T, Martinovic J, et al. Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney. Pediatr Nephrol, 2012, 27(1): 7-15
[25] Raychowdhury MK, Ramos AJ, Zhang P, et al. Vasopressin receptor-mediated functional signaling pathway in primary cilia of renal epithelial cells. Am J Physiol Renal Physiol, 2009, 296(1): F87-F97
