复旦大学附属儿科医院高通量测序数据分析流程(第二版)对遗传疾病候选变异基因筛选用时和准确性分析
2018-05-28董欣然彭小敏吴冰冰王慧君卢宇蓝周文浩
杨 琳 董欣然 彭小敏 陈 乡 吴冰冰 王慧君 卢宇蓝 周文浩,
全外显子测序(WES)是指对基因组DNA上所有蛋白质编码序列(外显子)进行序列检测和分析,临床外显子测序是指针对已知致病基因的全部编码区进行序列检测和分析。这2项技术均属于高通量测序的范畴。随着高通量测序技术在科研及临床的应用,越来越多的医生,认可这项新的技术有益于患儿的诊断及临床决策的制定[1-3]。
高通量测序技术用于临床的主要瓶颈在于快速、准确和自动化的数据分析。复旦大学附属儿科医院分子诊断中心(本中心)在2015年建立了高通量测序数据分析和临床诊断流程(简称复旦流程1.0)[4,5],应用于临床诊断不明病例,提升了对于遗传性疾病的分子诊断水平。随着送检病例的不断增加,本中心内部数据库的不断积累,改进并完善数据分析流程,引入从病例信息直接进行表型提取的系统,根据表型进行候选变异位点的自动分析及评估,形成了目前使用的本中心高通量测序数据分析和临床诊断流程(简称复旦流程2.0)。
本文以同一批新生儿行分子诊断病例,分别以复旦流程1.0和2.0行针对数据分析结果、流程总耗时和准确性的比较和分析,考察复旦流程2.0快速、准确和自动的进行大样本量的数据分析的水平。
1 方法
1.1 病例纳入标准 2017年11月7~14日取得家属的知情同意的、送本中心进行临床外显子检测的连续病例。
1.2 考察指标 以复旦流程1.0和2.0对纳入病例针对数据分析结果、流程用时和准确性的比较和分析。
1.3 复旦流程2.0版本 图1显示,复旦流程2.0版本的5个功能,①临床关键信息抓取、分析和校验(浅黄色),②样本管理、测序实验和质控(浅绿色);③表型和测序数据汇总和更新(浅蓝色),④变异位点自动化注释和自动化分级系统(浅蓝色),⑤自动报告生成(浅蓝色)。
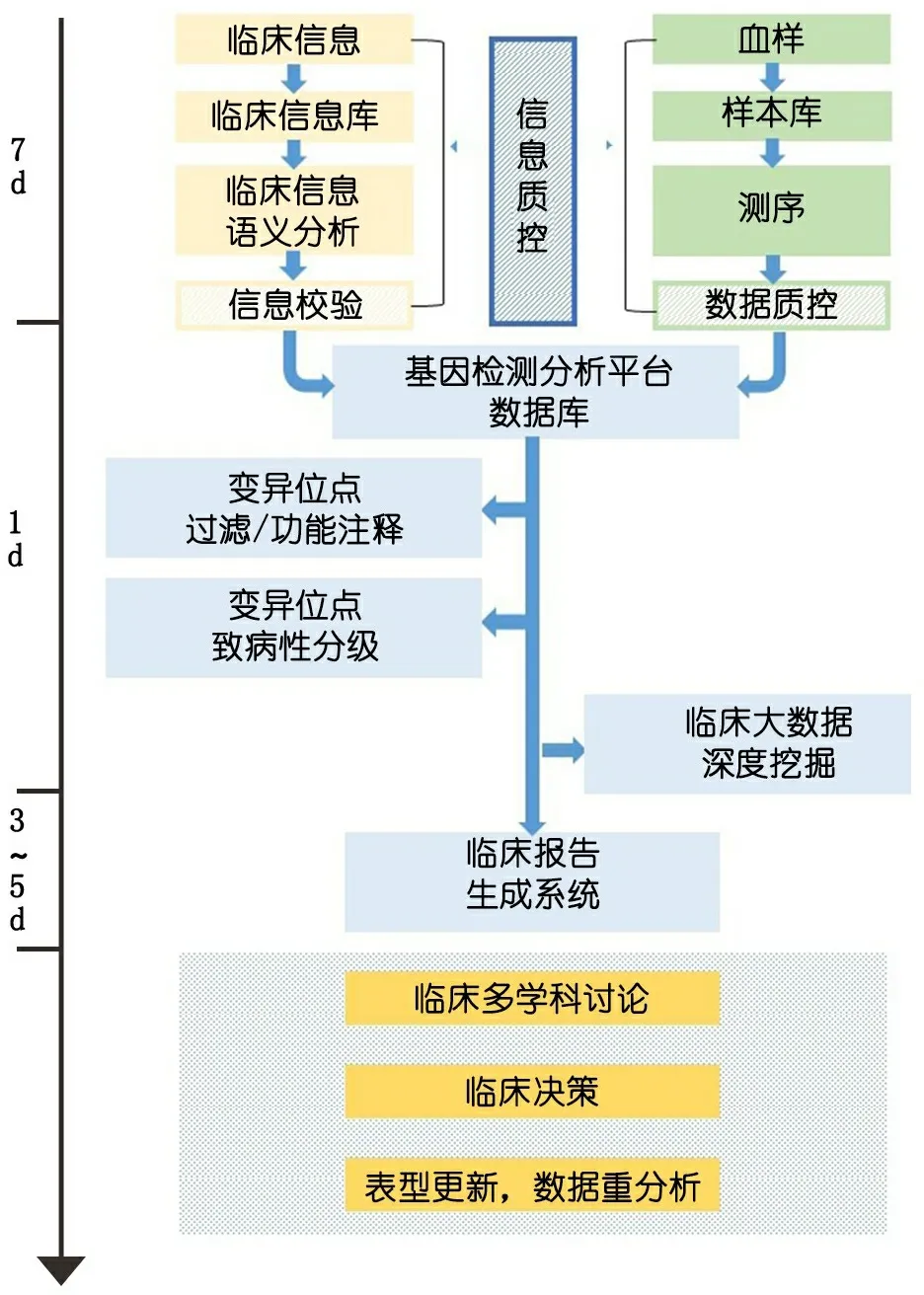
图1 复旦流程2.0版本功能示意图
1.3.1 临床关键信息抓取、分析和校验 临床信息自动化处理系统包含了3个主要工具模块。①分词模块,对原有的中文文本进行分词。将临床描述和诊断根据常见停顿符号分为短句和短语;英文的分词,采用UMLS提供的MetaMap软件[6]进行操作。②翻译模块,采用有道词典应用程序编程接口(API)的方式,将短语自动翻译为英文或者将英文再翻译回中文并将结果和现有语义库比对,如存在模糊匹配,在输出结果的同时将内容输入更新模块,由临床专家进行判定,如果匹配正确,存入中英文语义库。③临床信息自动化提取系统,包含中英文临床语义库和标准语义库(HPO),HPO数据库是将所有遗传相关表型进行标准化命名的网站[7]。临床语义库的词条都将和HPO中的术语进行关联,从而转换为结构化HPO术语。现有语义库通过专家校验增添中文语义537条,包含英文缩写DDH(HP:0001374 先天性髋关节脱位)等,中文缩写甲减(HP:0000851 先天性甲状腺功能减退症)等,以及皮肤黄染(HP:0000952 黄疸)、吃奶差(HP:0011968 喂养困难)等对症状的通俗描述。通过翻译最终匹配到英文语义库的中文短语也将进行记录,再次处理相同短语时将加快速度,现有此类条目关联共计57 667条,由于UMLS数据库比HPO数据库的条目多很多,因此其中48 867条并未最终匹配到HPO术语。
平台模块可以由临床病历提取系统自动触发,也提供了可视化交互界面,方便临床分子遗传学医生对语义库进行增删修改,并且对每个条目进行溯源。图2为复旦流程2.0平台模块临床信息自动化提取系统示意图,如原有的语句为“早产气促2天入院”,系统提取气促和早产2个关键词。早产为语义库中已有条目,直接输出对应的HPO术语HP:0001622(早产);气促并未匹配现有术语,但为病历高频词,由专家添加HPO术语HP:0002098(呼吸窘迫)关联至临床中文语义库;“极低出生体重儿”会匹配为HPO术语HP:0001518(足月小样儿)。随着语义库的持续定期更新,语义库将逐渐涵盖新生儿场景的所有表型信息。
1.3.2 样本管理、测序实验和质控 采用QIAGEN公司mini blood全血试剂盒及其标准DNA抽提方法提取基因组DNA(gDNA),用美国Thermofisher公司生产的NanoDrop紫外光分光度仪测定样本的浓度及定量。参照ClearSeq捕获试剂盒说明书,基因组DNA经过超声打断、末端修复、接头连接、杂交捕获。捕获文库采用Illumina HiSeq2000平台,进行序列检测。原始图像文件经Illumina base calling Software 1.7进行图像识别(Base calling),去除污染及接头序列处理后。Clean reads采用Burrows-Wheeler Aligner(BWA)软件v.0.5.9-r16,以人类基因组hg19(GRCh37)为参考序列进行比对。


图2 复旦流程2.0临床信息自动化提取系统
表型和测序数据汇总和更新引入了HPO数据库后,开发了临床信息自动化处理系统,实现了将HIS系统中病例文书的内容进行自动化抽提和分析,进而得到HPO术语(图2)。在复旦流程2.0中,抽提的病例文书包含出院小结、病程记录和门诊记录等,提取其中的临床诊断和描述,优先级为出院诊断>入院诊断>门诊诊断>病程描述。
1.3.4 变异位点自动化注释和自动化分级系统 在复旦流程1.0版基础上精简了筛选评估逻辑,升级拓展了本地人群数据库和公共人群数据库,添加了部分疾病和相关症状的中文描述,并增加了外显率、发病年龄和疾病系统。包括以下7个主要步骤。
1.3.4.1 质量控制 主要核对并解析所输入VCF文件的版本和格式,合并SNV和Indel的结果,临时拆分多态性位点,并精简出后续分析所关注的测序相关质量控制信息。关键信息包括变异dbSNP编号、变异的覆盖深度、符合参考碱基和符合变异碱基的测序片段数目、位点的质量得分和分级、以及初步的基因型判断(纯合或杂合)。这一步视测序质量,实践中一般滤去0~1个位点。
1.3.4.2 捕获测序区域筛选 基于捕获的二代测序(如全外显子组测序或基因panel),理论上所检测到的变异应该都位于捕获区域。但由于实验或分析方面的误差,以及患儿基因组上的特殊情况,VCF中往往含有捕获区域之外的变异。这些变异很可能为实验误差,但也有可能为真实的致病变异。考虑到后续分析往往围绕既往报道史和变异对基因功能的影响来展开,这一步筛选将会筛除所有距离外显子区15bp以外的变异,但既往文献报道致病的突变不受影响。这一过程与VCF的生成过程有关,实践中一般会滤去约15 000个位点。
1.3.4.3 公共人群频率注释筛选 引入gnomAD数据库(http://gnomad.broadinstitu te.org/),增加公共人群频率参考来源。该数据库包含8 624份东亚人群的外显子测序数据和811分东亚人群的全基因组测序数据。在实际操作过程中,会滤掉7 000~10 000个位点。
1.3.4.4 本地人群频率注释筛选 根据不同的测序捕获技术(WES或各种Panel)建立本地人群子库,更加精确的区分测序平台误差变异和人群高频位点;建立基因“白名单”和“黑名单”系统,保护人群高频的已知致病/功能性多态位点不被筛选(白名单),以及排除因假基因/异常结构导致测序错误的假阳性位点的干扰(黑名单);通过按染色体分表等方式改进数据库查询效率,综合提升分析速度。会滤去800-1 000个位点。
1.3.4.5 危害性注释筛选 与复旦流程V1.0并相似,仅对ANNOVAR和VEP的数据库版本进行了更新,并改善了关键信息的提取(比如不同转录本的选择、涉及重叠基因时的基因优选等)。但由于流程本身涉及人群频率和既往报道史的综合判断,所以整体分析结果得到了进一步改善。滤去的位点80~100个。
1.3.4.6 遗传模式优选 在复旦流程V1.0的基础上,增加了对外显率不全的考虑,使得家系分析中对于显性基因的判断更为灵活。与美国贝勒医学院合作,在OMIM数据库的基础上进一步参考美国贝勒医学院的遗传模式补充记录,丰富已知致病基因可能的遗传模式。滤去约20个位点。
复旦流程2.0的分析系统引入了表型关联分析和临床报告精选2项新的功能。表型相关分析基于先验的贝叶斯模型,同时考虑表型的特异度和疾病的表型丰富度,能更好地找出满足关键症状的相关基因(该算法的方法学文章待发表)。临床报告精选系统基于ACMG的指导框架,运用机器学习手段综合考虑变异危害程度、人群频率、既往报道、遗传模式等特征值,精选出最需要优先考量的致病位点,大大加快了临床专家对于致病性位点的寻找和判定。
1.3.4.7 变异分级的判断标准 对每个病例,系统分析得到的临床精选位点约15个,表型相关位点考虑排名前5的基因。若临床精选位点同时也满足表型相关,则列为主要发现候选;若表型关联较弱,则列为其他发现候选;若表型关联较高的位点不满足临床精选的标准,则列为相关发现候选。这种变异分级是在ACMG的变异五类分级的基础上,针对具体的临床干预措施而进一步区分。从ACMG变异分级指导结果来看,最终纳入报告候选的变异仅包括致病、可能致病和意义不明三类位点。从报告的可读性和精简性出发,一方面报告中不再罗列良性和可能良性的变异位点;另一方面报告直接结合患儿送检时的临床表型记录,将具有致病潜质的变异进一步分类为主要发现、相关发现和其他发现。其中,主要发现中列出了符合当前患儿临床表型的明确致病变异。这一类变异具有很高的临床诊断价值,应高度重视并参考;相关发现中列出的则是与当前患儿临床表型相符,但致病性不够明确的变异,例如不符合遗传模式或意义未明的错义突变。这一类变异应当进一步完善家系数据,确认疾病的遗传模式或是采用其他检测方法进一步挖掘潜在的变异(例如LOH、父母生殖细胞嵌合等情况);其他发现中则列出了致病性较为明确或是有既往报道史,但当前患儿并未体现出相关临床症状的变异位点。这一类变异建议临床考虑进一步丰富明确患儿症状,或是在未来的随访中高度关注变异相关的表型。
1.4 自动报告生成 基因检测报告从内容上不仅仅是罗列致病位点,还需要包含致病位点的解读说明等;从操作上,在形成规范化报告文档的同时需要避免冗余的重复性操作。临床快速报告自动化生成系统,选取文档模板,自动从平台数据库抽提患者基本信息形成报告表头;根据位点标注分级结果,给出结论,并将所选位点填入对应表格(主要发现、次要发现和补充发现),并且选取HGVS标准化命名、自动补充该位点的染色体位置、OMIM注释、HGMD注释、父母来源等信息;根据位点所在基因信息,自动补全对该基因的生物学功能描述,这些描述存在平台内部数据库中,提供了可视化平台对描述进行添加、更新等操作。在现有数据库平台中,已经包含683条对基因的描述信息,每条描述包含了对该基因关联的OMIM疾病概述、基因的致病机制、该疾病的主要临床表现以及遗传模式、疾病发作时期等信息。报告自动化系统的可视化平台还包含了多种辅助功能,可以快速提交Sanger验证、多报告汇总、报告加密、报告归档等请求。
变异评级标准参考了美国遗传学会的变异评级标准[8],制定本中心变异评级标准。
致病变异的标准为,①与先证者表型相符;②为已经明确的致病变异。
可疑致病变异的标准为,①与先证者表型相符;②与已经明确的致病变异有相同的氨基酸改变(不同的碱基改变);有害变异(无义变异,移码变异,典型+/-1或2剪接位点变异,起始密码子变异,单个或多个外显子缺失),且该基因功能缺失为已知的致病机制;③符合该致病基因已知的遗传模式。
相关变异的标准为,①位点所在基因的相关疾病与目前患儿表型部分相关;②符合显性遗传模式的临床意义不明确的遗传变异;③符合隐性遗传模式,仅发现一个致病/疑似致病的杂合致病变异;④符合隐性遗传模式,发现纯合的或复合杂合的临床意义不明确的变异。
其他发现的标准为,位点所在基因的相关疾病与目前患儿表型部分不相关;同1.3.3中②;符合该致病基因已知的遗传模式。
2 结果
2.1 一般情况 符合本文纳入标准112例患儿进入分析,男性50例,女性62例。年龄在10 h至28 d。复旦流程1.0和2.0,均由生物信息专业资深成员建立及维护,人工审核由经过临床遗传学培训的临床医生完成。该团队每年完成超过5 000例的高通量测序临床报告的分析及解读。
2.2 结果的比较 复旦流程1.0包括获得测序原始数据、拼接连接比对、获得变异结果、变异注释、生物信息学筛选、人工数据分析及报告书写等7个主要步骤。复旦流程2.0,在变异经过生物信息学筛选后,通过病例中提取临床表型,自动表型基因型比对流程,即通过表型对于变异进行逐个评级,将致病变异及相关变异分别列出。数据分析结果显示(表1),复旦流程2.0较复旦流程1.0在升级了变异的注释筛选、整合了表型进入筛选后,需要人工进行判读的变异数量大幅度的减少。使得高通量数据分析的时间得以压缩,提高了数据分析的效率。
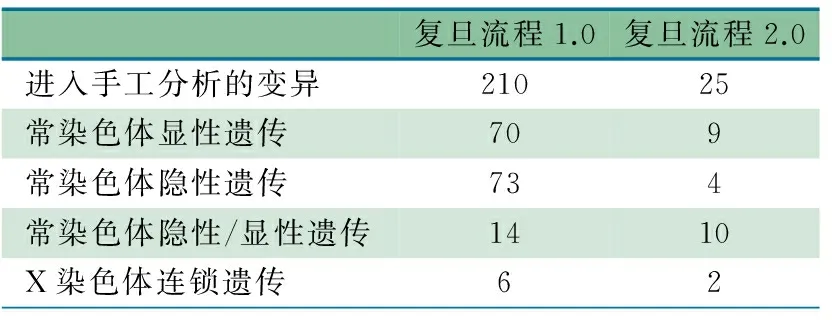
表1 复旦流程1.0与2.0进入手工分析变异的比较/个
2.3 用时比较 复旦流程1.0和2.0完成112例从样本送达到初步报告形成的时间,分别为78.8 h和19.8 h。表2显示,HPO的提取、变异的分级和报告的撰写,复旦流程1.0和2.0分别为78.8 h(4 725 min)和19.8 h(1 186 min)。

表2 复旦流程1.0与2.0每个步骤所用时间的比较
本文112例样本口头报告时间为13~16 d,报告发出的总TAT为20~33 d。测序环节(从收到样本到DNA提取、建库、上机测序)2~3周;数据分析到报告撰写环节,利用已经建立好的流程2.0,达到了在测序数据拿到24h内,口头报告阳性病例。
2.4 准确性的比较 112例经人工审核,8例检测到致病/可疑变异,12例检测到相关发现,其中3例经家系验证后,升级为致病/可疑变异,阳性率为9.8%(11/112)。复旦流程2.0与人工审核结果判读符合率82.1%(92/112),其中与人工审核后阳性结果判读符合率为63.6%(7/11),与人工审核后阴性结果判读符合率84.2%(85/101)。

图3 复旦流程2.0对1例维生素B6依赖性癫患儿致病变异的锁定
3 讨论
3.1 NGS数据分析的发展过程 目前国际上NGS的大型数据中心有:美国的Genome Center at White Head/MIT、Washington University Genome Center、Genome Center at Baylor Medical Collage、英国的Sanger Center,中国的华大基因研究中心等。成规模的数据分析的中心有:Baylor Medical Collage、GeneDex、UCLA、Ambry Genetics等。其中,Baylor Medical Collage的Genome Center成功建立了将基因组水平的变异分析用于临床分子的诊断的经典高通量测序数据分析流程[2,3]。
Stephen Francis Kingsmore教授等所在的美国Children’s Mercy Hospital的儿科基因组学中心及University of Missouri-Kansas City医学院,该研究团队通过表型-基因型关联数据库(Symptom-and sign-assisted genome analysis correlation tool, SSAGA)、HiSeq 2500测序、基因组序列比对软件、变异快速分析解释工具(Rapid Understanding of Nucleotide variant Effect Software, RUNES),可以在50 h做到从NICU患者血样DNA的提取到WGS数据分析解释工作全部完成[6],并且有研究报道已经到达了26 h。
上述研究及临床应用,均为高通量测序数据分析流程真正用于临床打下了坚实的基础。本中心在2014年,通过与Baylor Medical Collage的合作,初步形成了复旦流程1.0,并用于临床诊断[9]。2014至2015年每周的行高通量测序的样本量10~30例。随着NGS成本的不断下降及临床医生对于该项技术的认可度不断提升,现在每周样本量100~200例,需要提供更加快速、准确的数据分析流程,用以更大范围的解决临床问题。
3.2 复旦流程2.0的主要优势 ①从病历中自动提取先证者和/或核心家系的临床信息。复旦流程1.0中,临床信息的提取完全依靠手工,每个病例从住院病史及门诊病例中,整理需要的相关信息,并从中总结出主要的临床表型,用于下一步的表型基因型关联性分析,每个病例的平均耗时10 min。复旦流程2.0引入了临床信息自动化处理系统。通过分词模块、翻译模块,完成临床信息的初步提取,将提取出的信息和语义库进行比对,标注成为标准格式的HPO term。这些HPO term可以直接用于后续自动化的表型基因型关联性分析。②自动化表型基因型比对流程,即根据表型对于变异进行逐个评级,将致病变异及相关变异自动列出。这一功能的实现,主要依靠公共数据库中(OMIM、HGMD、HPO等),已经明确的基因与表型之间的关系,将每个基因对应到相关的多个表型。一旦患儿的HPO term出现该表型,特定的基因便会自动列为候选基因,该基因上的特定类型的变异就作为致病/可疑致病变异。③内部数据库的不断扩大,对于致病变异及良性变异的数量成指数级增加。本中心高通量基因检测数据库中,已经纳入的样本数超过15 000例。内部数据库的完善,可以快速明确注释致病变异及良性变异。为复旦流程2.0的建立也提供了坚实的基础。通过内部数据库的建立,对于表型的常见遗传病因谱及特定基因的热点突变谱,形成了检测人群特异性的数据。
3.3 复旦流程2.0后续的改进方向 人工智能使得对于海量数据的整理和管理成为可能。基因组水平的数据分析更是需要依靠自动化的“智能”流程,才能实现快速、准确。复旦流程2.0与人工审核后阳性结果判读符合率为63.6%(7/11),与人工审核后阴性结果判读符合率84.2%(85/101)。说明目前还不能完全依靠机器判读,必需结合人工判读,特别是对致病/可疑致病的判读。随着数据库的不断丰富和更新,加之引入机器学习来进一步扩充表型基因型的关联性,机器判读不致病/不可疑致病能力会进一步优先提高,同样也会带来对于变异位点的自动判读,尤其对于高度异质性的疾病,自动数据分析准确性的提高。随着人工智能技术所占比例的不断增加,表型基因型关联性的建立更加智能,逐渐减少人工数据分析及审核所占的比重。
目前复旦流程2.0中表型基因型的关联性,主要依靠公共数据库中(OMIM、HGMD、HPO等)已经明确的基因型与表型之间的关系。上述公共数据库中总结的患者表型,包含亚洲人、中国人数据相对较少。本中心高通量基因检测数据库中,已经纳入的样本数超过15 000例。通过内部数据库完善,建立内部的表型基因型关联性,使得复旦流程2.0更加适合中国特定人群的数据分析。
致谢:衷心感谢对本中心高通量基因检测数据库做出贡献的患儿及其家属,这些重要的贡献不仅对中国人群、同样对世界人群基因与表型关系都是弥足珍贵的,复旦流程2.0将继续努力不辜负你们的贡献。
参考文献
[1] Miller NA, Farrow EG, Gibson M, et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med, 2015, 7: 100
[2] Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med, 2013, 369(16): 1502-1511
[3] Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA, 2014, 312(18): 1870-1879
[4] 黎籽秀, 刘博, 徐凌丽, 等. 高通量测序数据分析和临床诊断流程的解读. 中国循证儿科杂志,2015,10(1):19-24
[5] 黎籽秀, 刘博, 杨琳, 等. 高通量测序数据分析和临床诊断流程对新生儿多发畸形候选变异的筛选准确性研究. 中国循证儿科杂志, 2015, 10(1): 25-28
[6] Saunders CJ, Miller NA, Soden SE, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med, 2012, 4(154): 154ra135
[7] Kohler S, Vasilevsky NA, Engelstad M, et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res, 2017, 45(D1): D865-D876
[8] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015, 17(5): 405-424
[9] 杨琳, 黎籽秀, 梅枚, 等. 全外显子组序列分析新生儿FGFR2基因相关疾病1例. 中国循证儿科杂志, 2015, 10(1): 34-39
[10] Mills PB, Footitt EJ, Mills KA, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain, 2010, 133(Pt 7): 2148-2159
[11] Been JV, Bok LA, Willemsen MA, et al. Mutations in theALDH7A1 gene cause pyridoxine-dependent seizures. Arq Neuropsiquiatr, 2008, 66(2A): 288, author reply 288-289
[12] Milh M, Pop A, Kanhai W, et al. Atypical pyridoxine-dependent epilepsy due to a pseudoexon inALDH7A1. Mol Genet Metab, 2012, 105(4): 684-686
[13] Yang Z, Yang X, Wu Y, et al. Clinical diagnosis, treatment, andALDH7A1 mutations in pyridoxine-dependent epilepsy in three Chinese infants. PloS one, 2014, 9(3): e92803
