Sm3+,Sr2+共掺杂对CeO2基电解质性能影响的密度泛函理论+U计算∗
2018-05-08陈美娜张蕾高慧颖宣言任俊峰林子敬
陈美娜张蕾高慧颖宣言任俊峰林子敬
1)(山东师范大学物理与电子科学学院,济南 250358)
2)(中国科学技术大学物理系,合肥 230026)
(2017年12月26日收到;2018年2月2日收到修改稿)
1 引 言
中温固体氧化物燃料电池(IT-SOFC)[1−8]是一种燃料普适性强、能量转化效率高,无贵金属催化剂且排放物无污染的新型发电设备.萤石结构的CeO2基电解质材料由于在500—700◦C(IT-SOFC的工作温度)下较传统的YSZ(氧化钇稳定的氧化锆)电解质材料表现出更强的氧离子传输性能(例如,600◦C下,YSZ的离子电导率为0.0032 S/cm,而Sm3+掺杂的CeO2材料离子电导率可达0.02 S/cm[7]),从而受到了人们的广泛关注.
实际上,纯相的CeO2材料在IT-SOFC的工作条件下,由于热激发产生的少量氧空位以及低氧分压下Ce4+到Ce3+的转变,是一种拥有极低的氧离子电导率和电子电导率的混合导体材料.而高性能的IT-SOFC电解质材料则要求材料具有较高的氧离子电导率以提高氧离子的传输性能以及越低越好的电子电导率以降低电解质的内短路电流.为了提高CeO2基材料的氧离子电导率,人们通常会在CeO2中掺入某种二价或者三价元素,通过电荷平衡来产生氧空位.近年来,关于CeO2的实验和理论研究有很多[9−19],人们发现在单掺杂体系中,掺杂离子的价态、半径及掺杂浓度等很多因素都能在某种程度上影响CeO2基材料的离子电导率[20−27].Nakayama和Martin[28]通过第一性原理计算的方法研究了Sc,Y,La,Nd,Sm,Gd,Dy及Lu等元素掺杂的CeO2基材料,发现体系的缺陷关联能和氧离子迁移能主要受到掺杂离子半径的影响.Shi等[22]利用PBE+U方法计算了Mn,Pr,Sn,Zr等元素掺杂的CeO2,发现掺杂引起的晶格畸变和电子结构变化是引发氧空位形成的两种主要机制.Yahiro等[29]通过实验研究证实,在镧系元素中,Sm3+或Gd3+单掺杂的CeO2(通常被称为SDC或GDC)呈现出最高的氧离子电导率,因此,近年来SDC和GDC在IT-SOFC领域得到了广泛的应用.Ou等[30]通过实验手段研究了Sm3+,Gd3+,Dy3+及Yb3+单掺杂的CeO2材料,他们认为氧空位的局域有序化是影响氧离子传输的主要因素,另外,他们还发现氧空位局域有序化的程度会随着掺杂离子的种类按照(Gd3+,Sm3+)>Dy3+>Yb3+的顺序以及掺杂离子浓度的增加而增大,这制约了CeO2基材料离子电导率的进一步提高.由此,他们建议通过控制掺杂元素的析出和多种元素共掺杂等手段控制氧空位局域有序化的形成,从而达到提高CeO2基材料离子电导率的目的.
两种或多种元素共掺杂的CeO2基材料成为近年来的研究热点[2,6,31−34]. Kashyap等[31]研究发现,与单掺杂Gd3+相比,Sr2+和Gd3+共掺杂可以将CeO2基材料的离子电导率从6.3×10−3S/cm提高到1.1×10−2S/cm.Jaiswal等[32]报道称,Sr2+和Sm3+共掺杂CeO2的离子电导率可达2.67×10−2S/cm,是同等情况下Sm3+单掺杂CeO2的两倍.Banerjee等[35]发现Ce0.8Sm0.05Ca0.15O2−δ体系在温度为700◦C时离子电导率可高达1.12×10−1S/cm,而且这种材料在还原性气氛下有良好的化学稳定性.Yamamura等[33]研究了二价离子Ca2+,La2+及Sr2+,La2+对CeO2的共掺杂效应,他们认为氧空位的局域有序性被破坏是共掺杂能显著提高CeO2基电解质离子电导率的主要原因.Ji等[34]通过扫描电子显微镜、拉曼光谱、交流阻抗谱等实验手段对Pr3+,Sm3+共掺杂的CeO2进行了研究,他们认为Pr3+,Sm3+的共掺杂能够改变晶界环境,产生更多的晶界和内表面,从而促进氧离子电导率的提高.Cioaterǎ[36]制备了Sr2+,Eu3+共掺杂的CeO2材料,通过对实验数据的分析,他们指出晶界上Sr的偏析是引发Sr2+,Eu3+共掺杂CeO2氧离子电导率升高的主要原因.Tao等[6]最近发现,In3+,Sm3+共掺杂的CeO2基电解质材料,除了氧离子电导率相较单掺杂的情况得到了显著提升之外,电子电导率也得到了有效抑制,这使得CeO2基电解质材料的性能得到进一步的提升.
虽然很多实验已经证实共掺杂对CeO2基材料的离子电导率具有协同提高效应,然而,并非所有的实验都支持这一结论.Kasse和Nino[37]发现Sm3+,Nd3+共掺杂CeO2的离子电导率介于Sm3+和Nd3+分别单掺杂CeO2的离子电导率之间.Yoshida等[38]在La3+,Y3+共掺杂的CeO2基材料中也发现了类似的共掺杂平均效应现象.因此,弄清楚共掺杂影响CeO2基材料电导率的微观机理具有重要意义.
与实验手段相比,现阶段针对CeO2基材料共掺杂的理论研究十分匮乏,所得到的结论也都相对较初步,且无法达成一致.Burbano等[39]使用分子动力学方法,在不考虑晶界效应、杂质析出、掺杂不均匀等情况的前提下,得出了Nd3+和Sm3+或Sc3+和La3+的共掺杂对CeO2基材料氧离子电导率的改变是平均效应而不是协同提高效应的结论.Andersson等[40]基于密度泛函理论(DFT),计算并分析了CeO2中镧系元素单掺杂对氧离子电导率的影响,它们还预测出Sm3+,Nd3+或Pr3+,Gd3+共掺杂CeO2的氧离子电导率会比相应单掺杂CeO2的氧离子电导率更高(即共掺杂产生协同效应).然而,这一理论预测却与后续Kasse和Nino[37]关于Sm3+和Nd3+共掺杂CeO2的实验结果相违背(平均效应),其深层原因有待进一步探究.Alaydrus等[41]利用第一性原理的方法研究了Sm3+和Gd3+共掺杂的CeO2体系,与Burbano等[37]的结论不同,它们发现在共掺杂体系中,两种不同的掺杂元素对氧空位的关联作用呈现出一种复杂的竞争关系,并不是简单的用相应的单掺杂效应直接叠加或者由单掺杂的平均效应可以描述的.
本文在当前共掺杂对CeO2电导率的作用机理尚不十分明确的前提下,利用第一性原理计算的DFT+U方法,对Sr2+和Sm3+共掺杂的CeO2体系进行了系统的计算研究,从掺杂材料的电子态密度、能带结构、氧空位形成能及氧空位迁移能等微观属性出发,解释实验现象,揭示了Sr2+和Sm3+共掺杂协同提高CeO2基IT-SOFC电解质性能的微观机理,这对进一步研发高性能的CeO2基IT-SOFC电解质材料具有十分重要的理论和实践意义.
2 计算方法
本文的计算是基于DFT的第一性原理计算方法,采用Vienna ab initio simuation package(VASP)软件包[42]完成.计算过程中考虑了自旋极化,并选用了缀加平面波方法[43]和广义梯度近似中的Perdew-Burke-Ernzerhof(PBE)交换泛函[44].平面波截断能为400 eV,能量自洽收敛条件为10−4eV.每个原子上的Hellmann-Feynmam力的收敛判据为0.02 eV/Å.采用了以Γ点为中心的2×2×2的k点网格进行布里渊区积分,高斯展宽为0.05 eV.考虑到传统的DFT方法对研究体系中Ce和Sm的4f电子之间的相互作用描述不足的问题,采用了DFT+U的方法(Ueff=U−J).其中Ce的U=5.0 eV,J=0 eV[45],Sm的U值和J值分别取8.0 eV和0.65 eV[46],Sr和O由于不含高角量子数的f电子和d电子,所以不需要加U.另外,在计算氧空位迁移的过渡态能垒时,选用了爬坡式弹性能带(climbing image nudged elastic band,CINEB)[47]的方法进行计算.
3 结果与讨论
3.1 晶体结构
CeO2的晶体结构属于立方晶系中的萤石结构.为了计算掺杂离子的影响,选用了2×2×2的超晶胞,其中包含了32个Ce,64个O,共96个原子.首先进行了晶格结构的弛豫,优化后的纯的CeO2的超晶胞的晶格常数为5.442 Å,与实验值5.412 Å[48]很接近,证明了本文数据的准确性.
为了模拟Sr2+和Sm3+共掺杂的CeO2,在超胞中利用一个Sm和一个Sr取代原来的Ce,去除一个O来模拟具有一个氧空位的共掺杂结构,并根据两个掺杂离子之间的间距由最近邻(用N表示)、次近邻(用NN表示)、次次近邻(用NNN表示)、到次次次近邻(用NNNN表示)以及掺杂离子与氧空位之间距离的不同(也是类似用N,NN,NNN及NNNN表示氧空位与掺杂离子之间的间距),将Sr2+和Sm3+共掺杂CeO2的结构分为M1,M2,M3,M4,M2-2,M3-2,M4-2,M5-2,M2-3及M4-3共10类.这些结构中掺杂离子与氧空位在超晶胞中的位置总览如图1所示.图2是标注了弛豫后能量的共掺杂局部结构示意图.例如,图2中M1结构指的是氧空位距离Sr2+和Sm3+是最近邻(N)且Sr2+和Sm3+之间也是最近邻(N)的构型,其结构弛豫后的能量为−772.75 eV,而M4-2结构则对应于氧空位距离Sm3+是次近邻(NN),距离Sr2+是最近邻(N)且Sr2+和Sm3+之间为次次近邻(NNN)的情况,其结构弛豫后的能量为−772.77 eV,图中其他掺杂结构的构型及能量皆可以此类推,在此不再赘述.
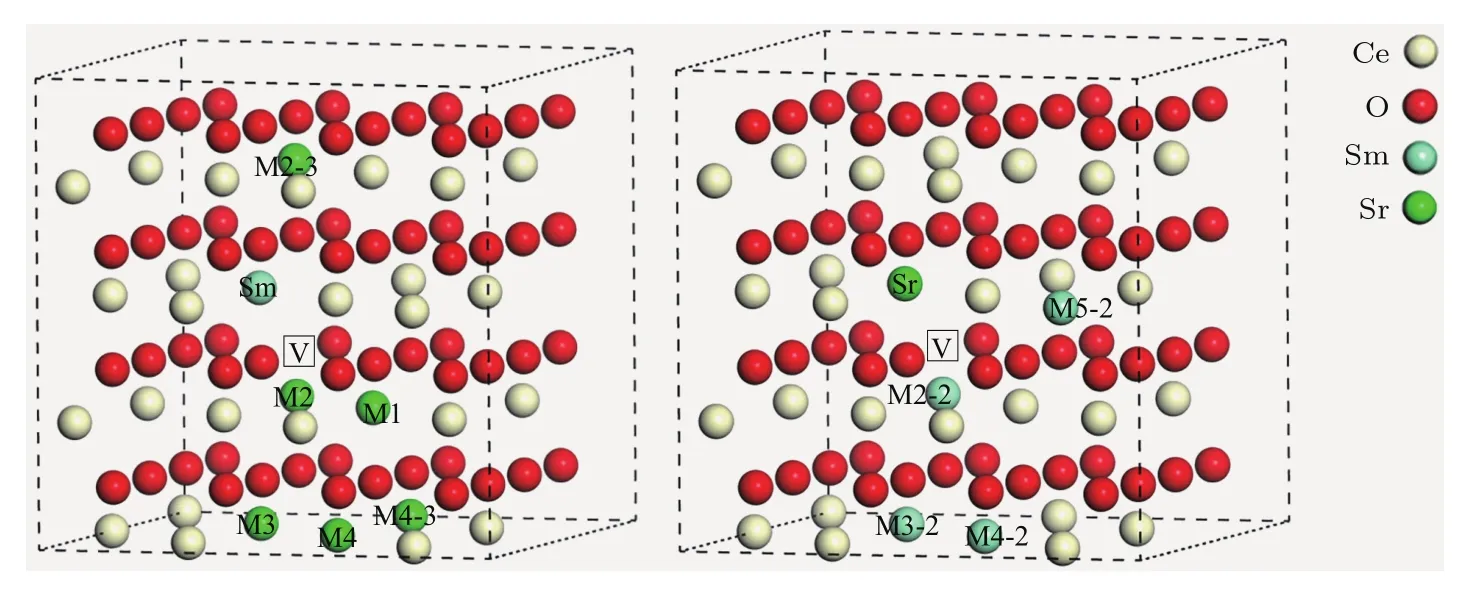
图1 掺杂离子与氧空位在超晶胞中的位置总览Fig.1.Location overview of the doped ions and oxygen vacancies in the supercell.
分析对比图2中M2,M3,M4三种掺杂构型,可以看到如果保持氧空位距离Sm3+最近邻,距离Sr2+次近邻不变,只改变两个掺杂离子Sm3+和Sr2+之间的距离,掺杂结构的能量会随着两掺杂离子之间距离的增大而降低.同理,分析M2-2,M3-2,M4-2这一列,能够得到相同的结论.但当两掺杂离子之间的距离进一步增大为NNNN时(由于此时两掺杂离子之间相距太远,没有办法找到氧空位距离一个掺杂离子最近邻、距离另外一个掺杂离子次近邻的构型,只找到了与前面几种结构构型最相近的M5-2),如图2所示.可以发现M5-2的能量比前面几种掺杂构型的能量都要高,这说明随着两掺杂离子之间距离的增大,掺杂结构的能量呈现出一种先降低后增高的变化趋势.这是由于共掺杂结构中掺杂离子与氧空位之间的库仑相互作用力与晶格畸变产生的应力相互竞争造成的.另外,通过分别比较M2和M2-2,M3和M3-2以及M4和M4-2这三组构型,发现当两掺杂离子之间间距不变时,氧空位距离Sm3+次近邻、距离Sr2+最近邻的构型,比氧空位距离Sm3+最近邻、距离Sr2+次近邻的能量要低.进一步比较M2和M2-3,M4和M4-3这两组构型,发现当保持两掺杂离子之间距离不变且氧空位距离Sm3+最近邻时,氧空位距离Sr2+次次近邻的构型比距离Sr2+次近邻的构型能量高.这说明在保持两掺杂离子之间距离不变的前提下,氧空位在不同位点的形成会影响体系的总能量.从另一个角度讲,保持两掺杂离子距离不变,共掺杂结构中氧空位在不同位点形成的难易程度是不同的,氧空位容易形成的位点体系的总能量会偏低.
综上,共掺杂CeO2体系的总能量会随着掺杂离子之间间距的不同和氧空位生成位点的不同而发生变化,且这种变化有一定的规律可循,这是由共掺杂结构中两种不同掺杂离子和氧空位之间的复杂相互作用造成的.
另外,由图2可见,当前计算的10种构型当中,能量最低共掺杂结构是M4-2和M1.因此,后续的工作将围绕着这两种能量最低的、结构具有代表性的共掺杂CeO2构型展开计算、分析和讨论.
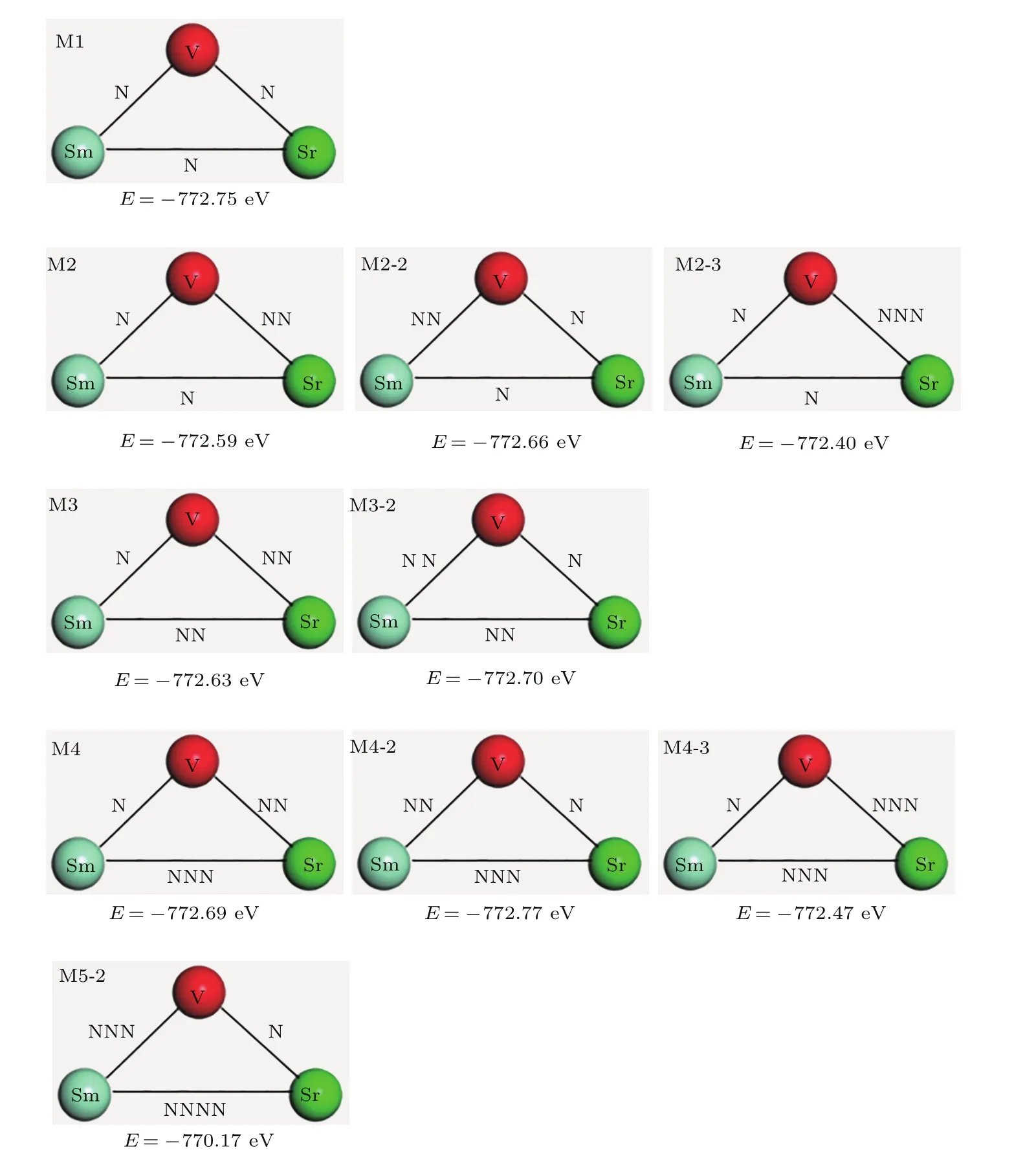
图2 共掺杂局部结构示意图及其对应能量Fig.2.Schematic diagram of the co-doped local structure and its corresponding energy.
3.2 电子态密度和能带分析
分别计算了完美结构的氧化铈(图3(a))、还原氧化铈(图3(b))、Sr2+单掺杂氧化铈(图3(c))、不同局域掺杂浓度的Sm3+单掺杂氧化铈(图3(d),图3(e))以及与M1和M4-2构型相对应的Sm3+和Sr2+共掺杂氧化铈(图3(f),图3(g))的电子态密度和能带结构(表1).
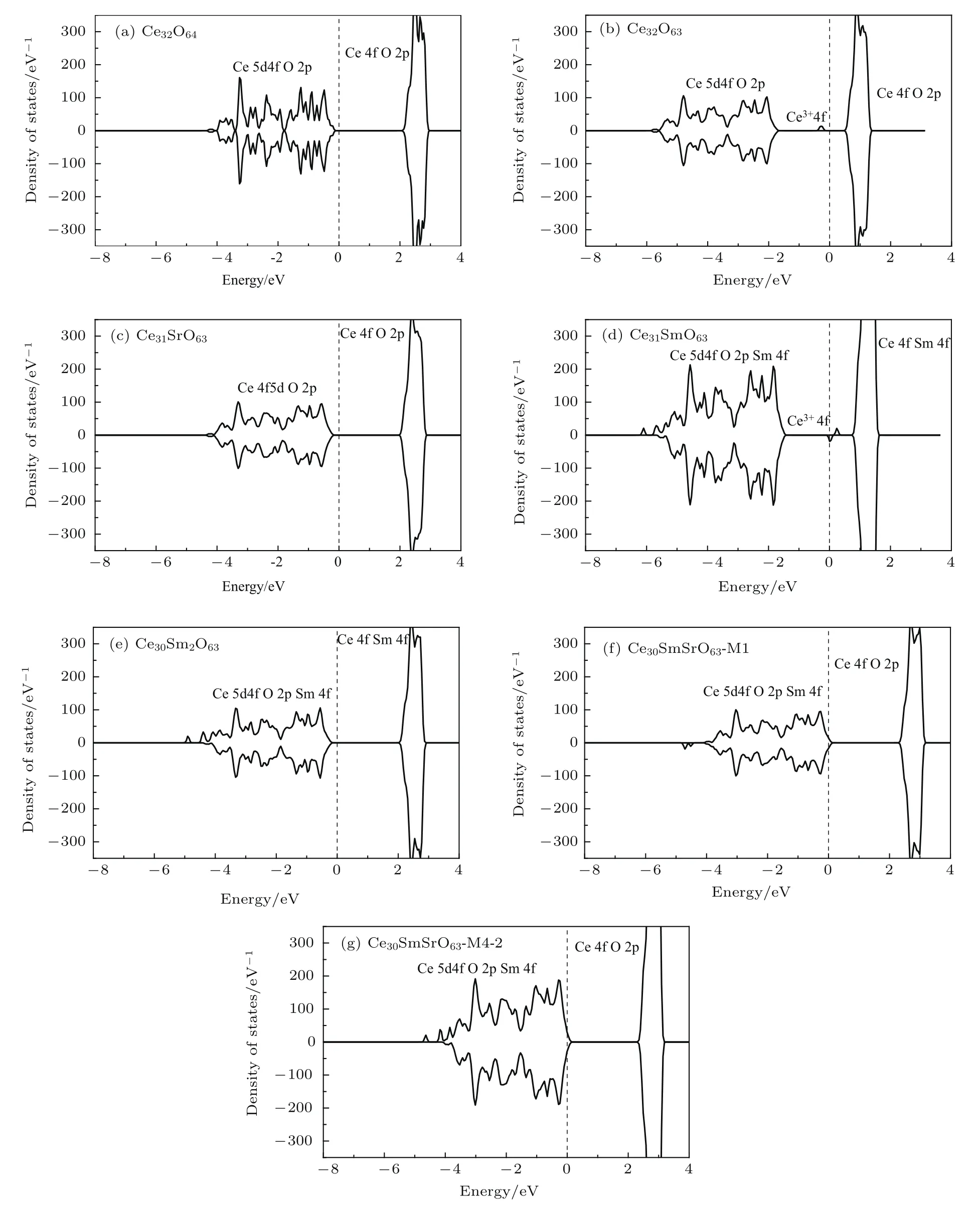
图3 Ce32O64,Ce32O63,Ce31SrO63,Ce31SmO63,Ce30Sm2O63(M1)和Ce30Sm2O63(M4-2)的态密度Fig.3.Density of states of Ce32O64,Ce32O63,Ce31SrO63,Ce31SmO63,Ce30Sm2O63(M1)and Ce30Sm2O63(M4-2).
结合图3(a)和表1可以看出,完美结构的氧化铈价带和导带相距较远,禁带宽度为2.372 eV,这说明完美结构的氧化铈电子导电性能不佳,为绝缘体.但当氧化铈晶体中由于热激发或者暴露在还原气氛中而产生氧空位时,如图3(b)所示,费米面附近会出现由Ce4+向Ce3+转变而产生的杂质能级,这会导致氧化铈的带隙降低到0.796 eV,从而能够很好地解释纯的氧化铈材料在还原性气氛或者高温下会有少量的电子电导率的原因.分析Sr2+单掺杂CeO2的电子态密度和禁带宽度(2.291 eV)可知,Sr2+的掺杂能够有效地抑制Ce4+到Ce3+的转变(没有杂质能级产生),而且带隙相对较宽,这说明Sr2+的掺杂有利于抑制电子电导率.
分析图3(d)和图3(e)可知,对Sm3+单掺的CeO2而言,是否产生Ce3+的杂质能级是由局域掺杂浓度决定的,当局域掺杂浓度较低时(Ce31SmO63),Sm3+周围的Ce4+会由于产生氧空位的电荷补偿机制转换成Ce3+,引发费米面附近杂质能级的产生,禁带宽度变窄(0.578 eV),并产生一定的电子电导率.而当局域掺杂浓度增高时(对应于Ce30Sm2O63),则由于刚好有足量的三价Sm3+参与氧空位的形成,抑制了Ce3+杂质能级的产生,相应的禁带宽度为2.316 eV,从而使得这种材料几乎不导电.而在实际的材料制备过程中,局域掺杂浓度实际上很难控制,这使得SDC中这两种局域掺杂情况(如图3(d)和图3(e))通常会共存,这就解释了SDC一般会有少量的电子电导率从而导致使用这种材料做电解质的IT-SOFC开路电压降低的原因.
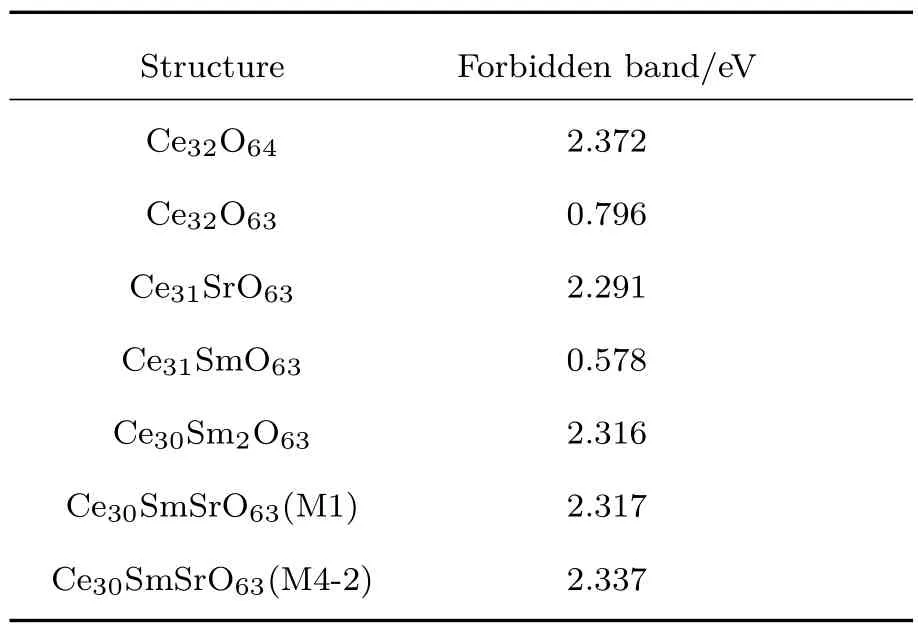
表1 纯相CeO2和掺杂CeO2的禁带宽度Table 1.Forbidden bandwidth of pure and doped CeO2.
结合图3(f)和图3(g)以及表1可以看出,无论是M1还是M4-2构型的Sm3+和Sr2+共掺杂的CeO2体系,都能很好地抑制Ce3+杂质能级的产生,且它们的禁带宽度(与M1对应的为2.317 eV,与M4-2对应的为2.337 eV)相较Sr2+或者Sm3+单掺杂的禁带宽度更宽,仅次于完美结构的氧化铈的禁带宽度.这说明Sm3+和Sr2+共掺杂的CeO2材料相较Sm3+或者Sr2+单掺杂的CeO2材料在抑制电子电导率方面更具优势,有希望成为性能更高的IT-SOFC电解质材料.
3.3 氧空位形成能
氧空位形成能是表征材料体系中氧空位形成难易程度的一个物理量,它对材料的氧离子电导率有着十分重要的影响.氧空位形成能的定义为从材料体系中去除一个氧所需的能量,公式为
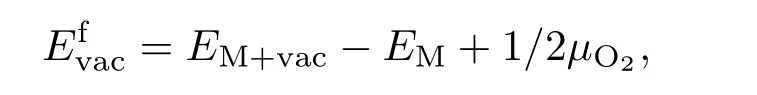
其中EM+vac是含有掺杂剂和氧空位的超胞的总能量,EM是含有掺杂剂不含氧空位的超胞的能量,µO2是O2的化学势.氧空位形成能如果是正值表示材料形成氧空位需要从外界吸收能量,氧空位形成能如果是负值则表示材料形成氧空位会向外界放出能量,亦即材料能够自发形成氧空位.
如表2所列,我们实际上计算了一系列CeO2基材料的氧空位形成能,这些材料包括还原氧化铈(用Ce32O63表示)、晶格中只有一个Ce4+被Sr2+或Sm3+取代的单掺杂氧化铈(分别用Ce31SrO63-N,Ce31SrO63-NN或Ce31SmO63-N,Ce31SmO63-NN表示,短横线后面的N和NN分别代表氧空位与掺杂离子是最近邻或次近邻)、晶格中有两个Ce4+被Sm3+取代且两个Sm3+之间为最近邻的Sm3+掺杂氧化铈(分别用Ce30Sm2O63(N)-N,N,Ce30Sm2O63(N)-N,NN表示,其中括号内的N代表两个掺杂Sm3+之间的间距为最近邻,短横线后的N,N代表氧空位距离两个Sm3+都是最近邻,而N,NN则代表氧空位距离其中一个Sm3+是最近邻,距离另外一个Sm3+是次近邻)以及Sm3+和Sr2+共掺杂的氧化铈(用Ce30SmSrO63(N)-N,N,Ce30SmSrO63(N)-NN,N和Ce30SmSrO63(NNN)-NN,N,Ce30SmSrO63(NNN)-NN,NN表示,其中括号里的N或NNN代表Sm3+和Sr2+之间为最近邻或次次近邻,而短横线后面的N,N,NN,N及NN,NN则分别代表氧空位距离Sm3+和Sr2+都是最近邻,氧空位距离Sm3+次近邻,距离Sr2+最近邻以及氧空位距离Sm3+,Sr2+都是次近邻,实际上,Ce30SmSrO63(N)-N,N对应于图2中的M1构型,而Ce30SmSrO63(NNN)-NN,N对应于图2中的M4-2构型).
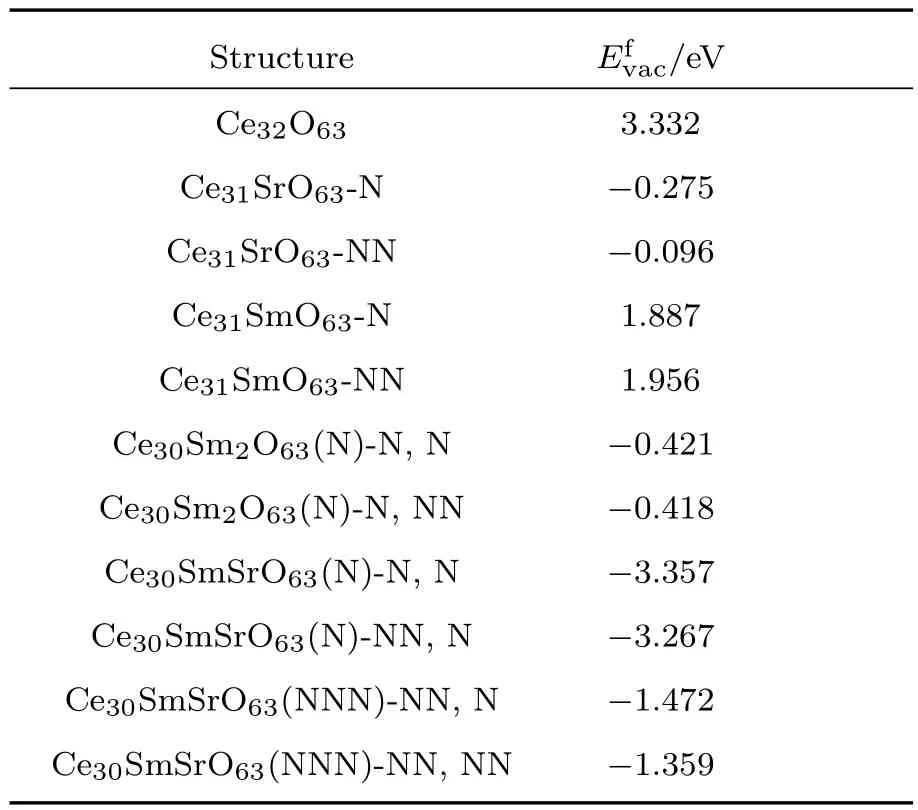
表2 CeO2基材料的氧空位形成能Table 2.Oxygen vacancy formation energy of CeO2based material.
由表2可以看出,纯的氧化铈Ce32O63的氧空位形成能很高,为3.332 eV,这说明纯的氧化铈很难形成氧空位,氧离子电导率很低.而Ce31SrO63-N,Ce31SrO63-NN的氧空位形成能分别为−0.275和−0.096 eV,差值为0.179 eV,这说明Sr2+的掺杂会使得CeO2能够自发形成氧空位,而且与Sr2+的次近邻相比氧空位更倾向于在Sr2+的最近邻处产生,这种现象同时也反映出Sr2+对氧空位有较强的俘获[49]能力,这种俘获能力会在一定程度上束缚氧空位的自由移动,因此,虽然Ce31SrO63有着很低的氧空位形成能,但由于Sr2+对氧空位的俘获效应,Ce31SrO63的离子电导率仍然会受到一定的影响.
对Sm3+掺杂的CeO2而言,Ce31SmO63-N,Ce31SmO63-NN的氧空位形成能分别为1.887和1.956 eV,而Ce30Sm2O63(N)-N,N,Ce30Sm2O63(N)-N,NN的氧空位形成能分别为−0.421和−0.418 eV,这说明Sm3+的局域掺杂浓度能够直接影响氧空位的生成,当Sm3+的局域掺杂浓度较低时(对应于构型Ce31SmO63),氧空位形成能为正值,不易形成氧空位,随着Sm3+掺杂浓度的升高(对应于构型Ce30Sm2O63),氧空位形成能变为负值,从而能够自发形成氧空位.另外,不同于Ce31SrO63所表现出来的杂质最近邻与次近邻位点之间氧空位形成能差距较大(约为0.179 eV)的现象,对Ce31SmO63而言,相应位点之间氧空位形成能的差值只有0.069 eV,说明Sm3+对氧空位的俘获效应没有Sr2+那么强,而对Ce30Sm2O63(N)而言,N,N和N,NN这两种不同位点之间的氧空位形成能差值进一步缩小,只有0.003 eV,这说明氧空位在这两种不同的位点处产生的概率近乎相等,这在一定程度上能够促进氧空位在不同位点之间的自由输运,这应该也是SDC通常具有比较高的离子电导率的原因之一.
进一步分析表2中的数据可知:Ce30SmSrO63(N)-N,N的氧空位形成能为−3.357 eV,Ce30Sm-SrO63(N)-NN,N的氧空位形成能为−3.267 eV,二者差值为0.09 eV,而Ce30SmSrO63(NNN)-NN,N的氧空位形成能为−1.472 eV,Ce30SmSrO63(NNN)-NN,NN的氧空位形成能为−1.359 eV,二者差值为0.113 eV.这一方面说明Sm3+和Sr2+共掺杂能够在Sm3+或Sr2+单掺杂CeO2的基础上协同降低氧空位形成能(Sm3+,Sr2+共掺杂的氧空位形成能低于相应的Sm3+或者Sr2+单掺杂的氧空位形成能,而不是介于Sm3+,Sr2+两种单掺杂材料的氧空位形成能之间);另一方面,由于Sm3+,Sr2+共掺杂的CeO2体系不同位点之间的氧空位形成能差值(0.09和0.113 eV)介于Ce30Sm2O63(N)的0.003 eV和Ce31SrO63的0.179 eV之间,说明Sm3+与Sr2+的共掺杂除了能够协同降低氧空位形成能之外,还能够部分中和Sr2+对氧空位较强的俘获效应,这两个方面实际上都有利于Sm3+,Sr2+共掺杂CeO2基材料离子电导率的提高.
3.4 氧空位迁移能
结合前面对晶格结构、电子态密度、能带结构以及氧空位形成能的分析,利用CI-NEB方法分别计算了Ce30SmSrO63(N)-N,N(对应于图2中双掺杂构型M1),Ce30SmSrO63(NNN)-NN,N(对应于图2中双掺杂构型M4-2)的氧空位向最近邻位置迁移(如图4所示,图中V代表氧空位,O1,O2,O3,O4代表不同的最近邻氧空位迁移位点)的过渡态能垒,为了与SDC做比较,我们还计算了SDC中Ce30Sm2O63(N)-N,N构型的氧空位向最近邻位置迁移的能垒,结果如表3所列.
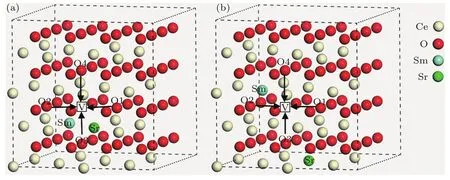
图4 共掺杂体系(a)M1和(b)M4-2的氧空位迁移路径Fig.4.Oxygen vacancy migration path of the co-doping system(a)M1 and(b)M4-2.
分析表3中M1构型的氧空位迁移能垒可知,氧空位迁移能垒按照M1-V→O3(N,N→N,N;Sm3+,Sr2+)>M1-V→O2(N,N→NN,N;Sr2+,Ce4+)>M1-V→O1(N,N→N,NN;Sm3+,Ce4+)>M1-V→O4(N,N→NN,NN;Ce4+,Ce4+)的顺序依次递减,又由于离子半径Sr2+(1.26 Å)>Sm3+(1.079 Å)>Ce4+(0.97 Å), 因此,经过分析发现,相较其他因素(例如,掺杂离子的库仑相互作用或是晶格畸变产生的应力)对氧空位迁移的影响,氧空位迁移路径边缘上的离子半径大小对氧空位迁移能垒的影响占据主导地位,大的离子半径会导致氧空位自由迁移空间的缩小,从而阻碍了氧空位的迁移,造成迁移能垒的增大.
对M4-2而言,迁移能垒按照M4-2-V→O1(NN,N→NNNN,N;Sr2+,Ce4+)>M4-2-V→O3(NN,N→NNN,N;Sr2+,Ce4+)>M4-2-V→O4(NN,N→NN,NN;Ce4+,Ce4+)>M4-2-V→O2(NN,N→N,NN;Ce4+,Ce4+)的顺序递减,由此可见,迁移路径边缘上的离子半径大小和两种不同掺杂离子对氧空位的相互作用共同主导了M4-2结构的氧空位迁移能垒.例如,当迁移路径边缘上的离子相同时,有M4-2-V→O1(NN,N→NNNN,N;Sr2+,Ce4+)>M4-2-V→O3(NN,N→NNN,N;Sr2+,Ce4+),且有M4-2-V→O4(NN,N→NN,NN;Ce4+,Ce4+)>M4-2-V→O2(NN,N→N,NN;Ce4+,Ce4+),这说明虽然氧空位在双掺杂氧化铈晶格中的迁移,会受到多种因素的影响,但就M4-2而言,固定迁移路径边缘上的离子相同且保持氧空位与Sr2+之间的距离不变,氧空位似乎更倾向于向距离Sm3+近的地方移动,这凸显出两种不同的掺杂离子对氧空位作用的竞争关系.
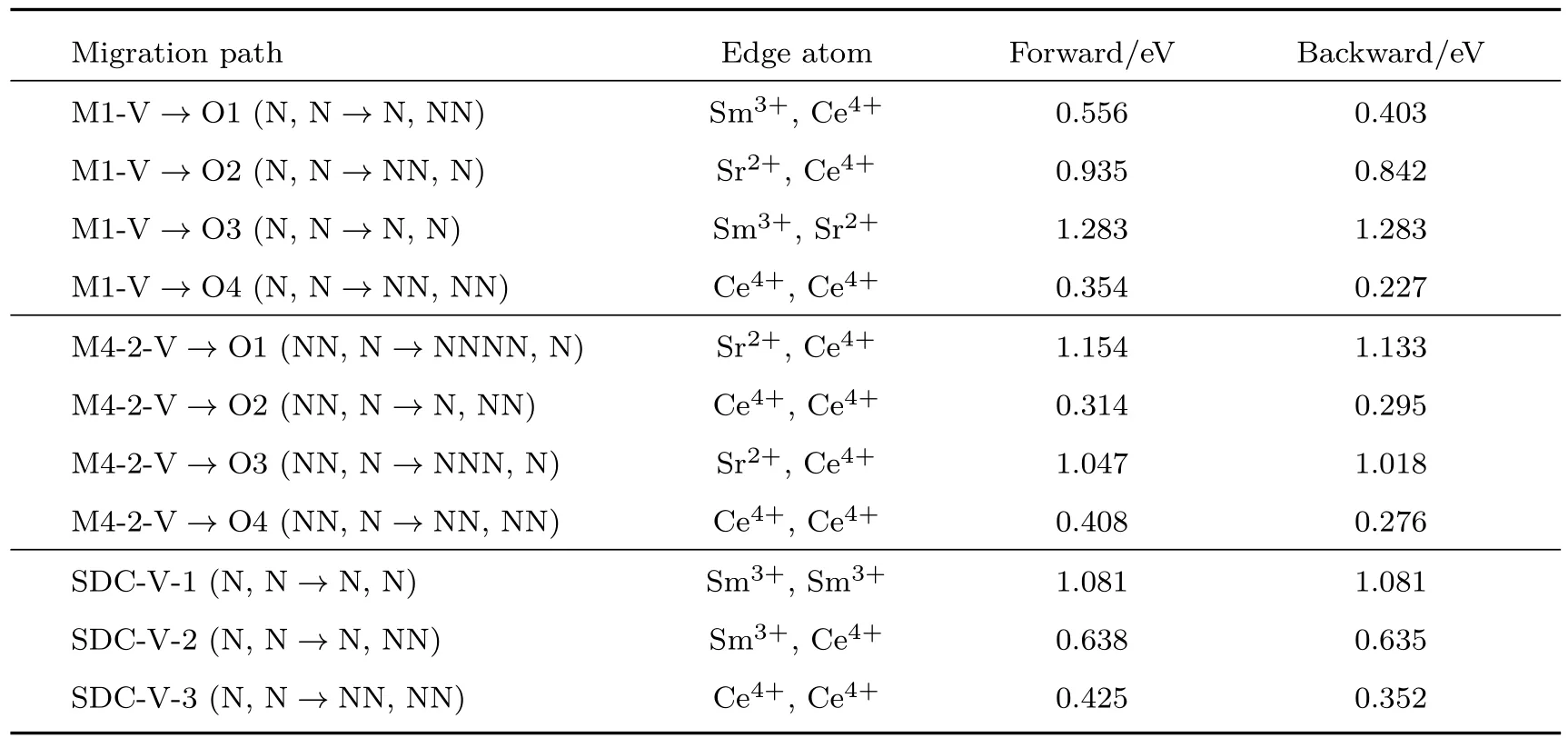
表3 M1,M4-2及SDC的氧空位迁移能Table 3.Oxygen vacancy migration energy of M1,M4-2 and SDC.
对比表3中M1和SDC相对应氧空位位点之间的迁移,发现当M1氧空位迁移路径边缘上有Sr2+出现时,M1-V→O2(N,N→NN,N;Sr2+,Ce4+)>SDC-V-2(N,N→N,NN;Sm3+,Ce4+),M1-V→O3(N,N→N,N;Sm3+,Sr2+)>SDCV-1(N,N→N,N;Sm3+,Sm3+),这意味着虽然氧空位是在M1与SDC相对应的位点之间迁移,但由于M1的迁移路径边缘上离子半径较大的Sr2+的出现,导致了M1的氧空位的迁移能垒大于SDC.而当M1的迁移径边缘上没有Sr2+出现时,M1和M4-2相对应位点上的氧空位迁移有:M1-V→O1(N,N→N,NN;Sm3+,Ce4+) 本文在共掺杂对CeO2电导率的作用机理尚不十分明确的前提下,利用第一性原理计算的DFT+U方法,对Sm3+和Sr2+共掺杂的CeO2进行了系统的研究,在初步筛选出Sr2+,Sm3+共掺杂CeO2体系最稳定构型的基础上,对比Sm3+或Sr2+单掺杂的CeO2体系,计算并分析了共掺杂体系的电子态密度、能带结构、氧空位形成能以及氧空位迁移能等微观属性.计算结果表明,Sm3+,Sr2+的共掺杂对CeO2基电解质性能的提高具有协同效应,二者的共掺杂不仅能协同抑制CeO2体系的电子电导率,还能在单掺杂CeO2的基础上进一步降低氧空位形成能,Sm3+的存在还有助于降低Sr2+对氧空位的俘获作用,而Sr2+的加入则能够在SDC的基础上进一步降低最低氧空位迁移能,爬坡式弹性能带方法计算表明共掺杂体系的氧空位迁移能最低可达0.314/0.295 eV,低于SDC的最低氧空位迁移能.本文的研究揭示了Sm3+,Sr2+共掺杂对CeO2电导率的协同作用机理,这对进一步研发其他高性能的共掺杂材料具有十分重要的理论和实践意义. 感谢中国科学技术大学超级计算中心的计算支持和帮助. [1]Steele B 2000Solid State Ionics129 95 [2]Maheshwari A,Wiemhöfer H D 2015Ceram.Int.41 9122 [3]Shi F 2010Int.J.Hydrogen Energ.35 10556 [4]Baqué L,Caneiro A,Moreno M S,Serquis A 2008Electrochem.Commun.10 1905 [5]Shi F,Song X P 2010Int.J.Hydrogen Energ.35 10620 [6]Tao Z T,Ding H P,Chen X H,Hou G H,Zhang Q F,Tang M,Gu W 2016J.Alloy.Compd.663 750 [7]Peng R R,Xia C R,Fu Q X,Meng G Y,Peng D K 2002Mater.Lett.56 1043 [8]Shi F,Xiao H T 2013Int.J.Hydrogen Energ.38 2318 [9]Chen L J,Tang Y H,Cui L X,Ouyang C Y,Shi S Q 2013J.Power Sources234 69 [10]Cui L X,Tang Y H,Zhang H,Hector Jr L G,Ouyang C Y,Shi S Q,Li H,Chen L 2012Chem.Chem.Phys.14 1923 [11]Shi S Q,Ke X Z,Ouyang C Y,Zhang H,Ding H C,Tang Y H,Zhou W W,Li P J,Lei M S,Tang W H 2009J.Power Sources194 830 [12]Shi S Q,Tang Y H,Ouyang C Y,Cui L X,Xin X G,Li P J,Zhou W W,Zhang H,Lei M S,Chen L Q 2010J.Phys.Chem.Solids71 788 [13]Tang Y H,Zhang H,Cui L X,Ouyang C Y,Shi S Q,Tang W H,Li H,Chen L Q 2012J.Power Sources197 28 [14]Li P J,Zhou W W,Tang Y H,Zhang H,Shi S Q 2010Acta Phys.Sin.59 3426(in Chinese)[李沛娟,周薇薇,唐元昊,张华,施思齐2010物理学报59 3426] [15]Bowman W J,Zhu J,Sharma R,Crozier P A 2015Solid State Ionics272 9 [16]Zha S W,Xia C R,Meng G Y 2003J.Power Sources115 44 [17]Nilsson J O,Vekilova O Y,Hellman O,Klarbring J,Simak S I,Skorodumova N V 2016Phys.Rev.B93 024102 [18]Guo C,Wei S X,Zhou S N,Zhang T,Wang Z J,Ng S P,Lu X P,Wu C M L,Guo W Y 2017ACS Appl.Mater.Inter.9 26107 [19]Tang Y H,Zhang H,Guan C M,Shen J Q,Shi S Q,Tang W H 2012Sci.Sin.-Phys.Mech.Astron.42 914(in Chinese)[唐元昊,张华,管春梅,沈静琴,施思齐,唐为华2012中国科学:物理学力学天文学42 914] [20]Fu Z M,Sun Q,Ma D W,Zhang N,An Y P,Yang Z X 2017Appl.Phys.Lett.111 023903 [21]Mogensen M,Sammes N M,Tompsett G A 2000Solid State Ionics129 63 [22]Tang Y H,Zhang H,Cui L X,Ouyang C Y,Shi S Q,Tang W H,Li H,Lee J S,Chen L Q 2010Phys.Rev.B82 125104 [23]Xiong Y P,Yamaji K,Horita T,Sakai N,Yokokawa H 2004J.Electrochem.Soc.151 A407 [24]Yoshida H,Inagaki T,Miura K,Inaba M,Ogumi Z 2003Solid State Ionics160 109 [25]Zhang D S,Qian Y L,Shi L Y,Mai H L,Gao R H,Zhang J P,Yu W J,Cao W G 2012Catal.Commun.26 164 [26]Zhang T S,Hing P,Huang H T,Kilner J 2002J.Mater.Sci.37 997 [27]Singh P,Hegde M 2010Cryst.Growth Des.10 2995 [28]Nakayama M,Martin M 2009Phys.Chem.Chem.Phys.11 3241 [29]Yahiro H,Eguchi K,Arai H 1989Solid State Ionics36 71 [30]Ou D R,Mori T,Ye F,Zou J,Auchterlonie G,Drennan J 2008Phys.Rev.B77 024108 [31]Kashyap D,Patro P K,Lenka R K,Mahata T,Sinha P K 2014Ceram.Int.40 11869 [32]Jaiswal N,Upadhyay S,Kumar D,Parkash O 2014Int.J.Hydrogen Energ.39 543 [33]Yamamura H,Katoh E,Ichikawa M,Kakinuma K,Mori T,Haneda H 2000Electrochemistry68 455 [34]Ji Y,Liu J,He T M,Wang J X,Su W H 2005J.Alloy.Compd.389 317 [35]Banerjee S,Devi P S,Topwal D,Mandal S,Menon K 2007Adv.Funct.Mater.17 2847 [36]Cioateră N,Parvulescu V,Rolle A,Vannier R 2009Solid State Ionics180 681 [37]Kasse R M,Nino J C 2013J.Alloy.Compd.575 399 [38]Yoshida H,Deguchi H,Miura K,Horiuchi M,Inagaki T 2001Solid State Ionics140 191 [39]Burbano M,Nadin S,Marrocchelli D,Salanne M,Watson G W 2014Phys.Chem.Chem.Phys.16 8320 [40]Andersson D A,Simak S I,Skorodumova N V,Abrikosov I A,Johansson B 2006Proc.Natl.Acad.Sci.USA103 3518 [41]Alaydrus M,Sakaue M,Aspera S M,Wungu T D,Linh T P,Kasai H,Ishihara T,Mohri T 2013J.Phys.:Condens.Mater.25 225401 [42]Kresse G,Furthmüller J 1996Phys.Rev.B54 11169 [43]Blöchl P E 1994Phys.Rev.B50 17953 [44]Perdew J P,Burke K,Ernzerhof M 1996Phys.Rev.Lett.77 3865 [45]Nolan M,Grigoleit S,Sayle D C,Parker S C,Watson G W 2005Surf.Sci.576 217 [46]Feng J,Xiao B,Wan C,Qu Z,Huang Z,Chen J,Zhou R,Pan W 2011Acta Mater.59 1742 [47]Henkelman G,Uberuaga B P,Jónsson H 2000J.Chem.Phys.113 9901 [48]Gerward L,Olsen J S,Petit L,Vaitheeswaran G,Kanchana V,Svane A 2005J.Alloy.Compd.400 56 [49]Lucid A K,Keating P R,Allen J P,Watson G W 2016J.Phys.Chem.C120 234304 结 论
