以帕金森综合征为主要表现的脊髓小脑性共济失调3型一例
2018-04-26马妍鲁明刘小璇樊东升刘晓鲁
马妍 鲁明 刘小璇 樊东升 刘晓鲁
脊髓小脑性共济失调(spinocerebellar ataxia,SCA)是一组高度遗传异质性疾病,包括多种亚型[1],我国以SCA3最常见,也称为马查多-约瑟夫病(Machado-Joseph disease,MJD)[2]。SCA3的核心表现包括为突眼、小脑性共济失调、肌强直、锥体束征、肌萎缩等,而以帕金森综合征为主要表现的SCA3病例较为少见。本文报道一例少见的以帕金森综合征为主要表现的SCA3病例。
1病例报道患者男性,39岁,主因“步态不稳伴肢体僵硬5年”于2016-01入北医三院神经内科就诊。患者于入院5年前无明显诱因出现行走及上下楼动作缓慢、僵硬,并逐渐加重,无明显波动性。入院1年前开始出现双手震颤,持物时明显,伴上肢活动迟缓,以右上肢为著,并自觉四肢肉跳,部位不固定,双足底疼痛。患者近1年来觉上下楼费力,行走缓慢、不稳,有前后倾倒倾向。发病以来未诉肢体麻木,无踩棉感,无头晕、复视,无言语不清或吞咽困难,嗅觉正常,大小便正常。其父50余岁开始出现步态不稳,步幅较大,无震颤、小碎步等表现,未诊治,67岁去世。内科检查:发育正常,肝脾无肿大;卧位血压122/64 mmHg,立位血压 121/75 mmHg。神经系统检查:神清语利,未见眼球突出,未见角膜K-F环,双眼各向活动充分,左右视可见水平眼震,无眼肌麻痹及面肌萎缩,伸舌居中,未见舌肌萎缩或束颤;双上肢肌张力齿轮样增高,双下肢肌张力铅管样增高,四肢肌力V级;双侧指鼻、跟膝胫试验稳准,反击征阴性;行走步态僵硬,协同动作少,步基稍宽,一字步(+),Romberg征睁闭眼均不稳;未见明显震颤及不自主运动,双侧轮替动作略笨拙,姿势反射异常;深浅感觉正常;除双侧跟腱反射未引出外,余肢体腱反射(++),双侧掌颌反射(+),右侧Chaddock征(+),左侧Chaddock征(±);简易精神状态检查表(MMSE)评分30分;蒙特利尔认知评估量表(MoCA)评分29分。实验室检查:微量元素(铜、锌、钙、镁、铁、铅)含量在正常参考范围;甲状腺功能、性激素、抗脑组织抗体、血清维生素B12、叶酸水平、抗核抗体(ANA)、抗可溶性抗原抗体(ENA)、抗中性粒细胞包浆抗体(ANCA)、抗双链DNA抗体(dsDNA)检测均在正常范围;血细胞镜检“未见棘红细胞”;免疫蛋白固定电泳“未见异常寡克隆区带”;血清铜蓝蛋白:45 mg/dL(正常参考值范围25~63 mg/dL);颅脑MRI未见明显异常(图1);腹部B超“未见肝脾肿大”;神经电生理检查:神经传导速度正常,左正中神经F波出现率下降,双胫神经H反射未测出,双下肢感觉诱发电位(SEP)中枢性损害,双侧上下肢运动诱发电(MEP)中枢性损害;左旋多巴负荷试验阴性;ATXN3基因检测“CAG重复序列杂合突变,数目分别为7和61”(图2)。患者CAG动态突变次数位于正常低限,临床易表现为锥体外系受累,考虑其父亲有小脑体征,且本患者对左旋多巴治疗无效,临床符合SCA3(4型)诊断。
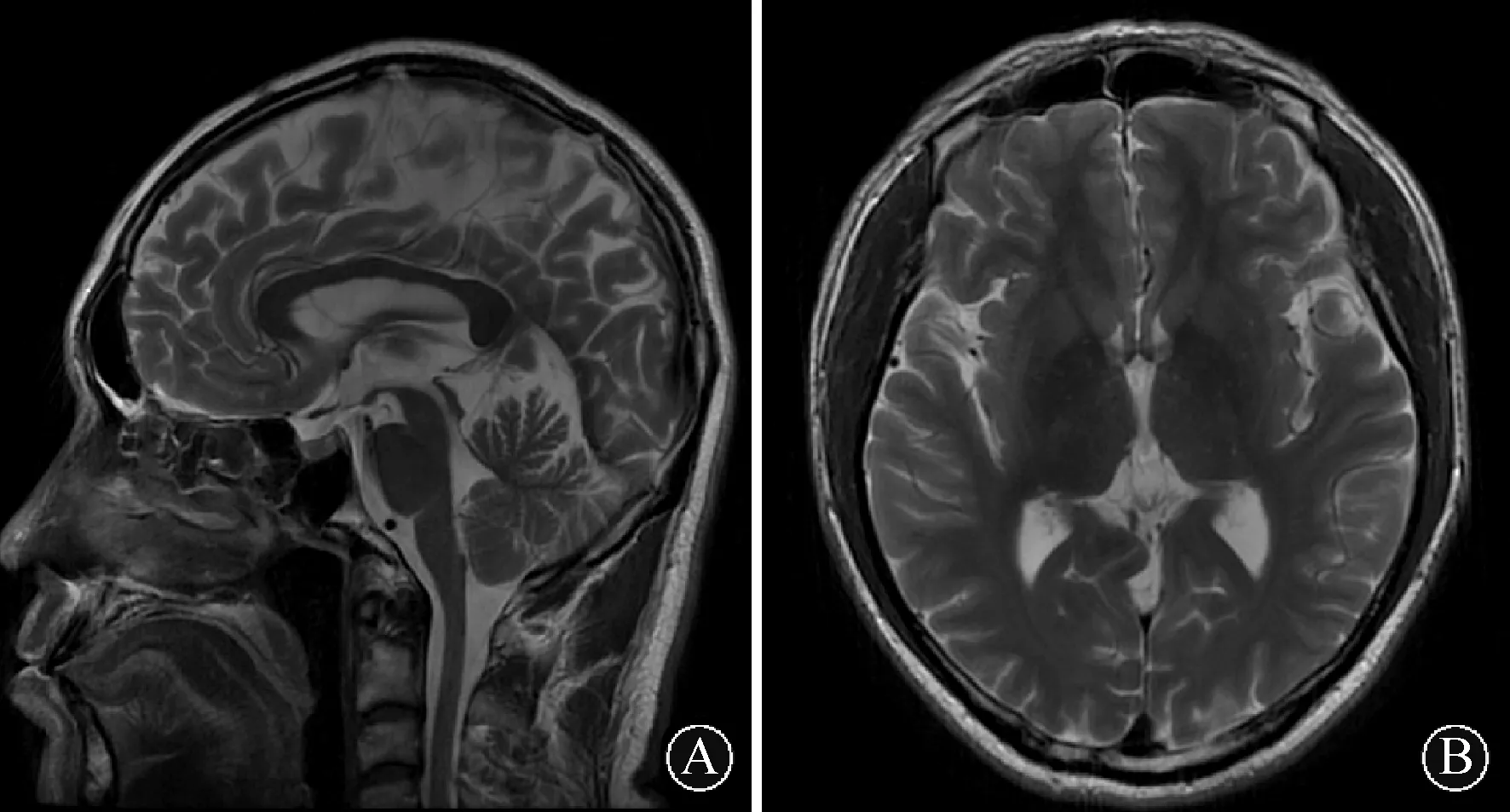
图 1 患者头MRI表现:患者颅脑MRI T2加权像矢状位未见大脑、小脑及脑干部位萎缩(A);患者颅脑MRI T2加权像横断位未见基底节区萎缩及异常信号(B)
2讨论SCA3是ATXN3基因CAG重复序列数目异常扩增所致,在中国人群中正常重复数为13~41次,异常者多大于60次[3]。SCA3的核心表现包括突眼、小脑性共济失调、肌强直、锥体束征、肌萎缩等。根据临床表现目前已报道5种亚型[4],其中4型发病晚,伴有帕金森综合征和周围神经病变。本患者临床以锥体外系受损为主,表现为帕金森综合征,虽有水平眼震、步基略宽等小脑体征及病理征,但指鼻、跟膝胫试验稳准,肢体反射不活跃,小脑体征、锥体束征相对较轻,且无SCA3常见的突眼、面舌肌萎缩等表现,临床表型较为少见。此外,患者有可疑家族史(其父有明确的小脑体征),这也提示SCA3诊断可能性大。
遗传学证实的SCA3患者合并锥体外系症状较少且多为个案报道[5],此症状在非洲裔患者中较多见[6]。锥体外系受累可有多种表现,包括肌张力障碍、静止性震颤、运动迟缓等。该类患者CAG重复次数通常属于异常范围的低限,对左旋多巴的反应不一。Maciel等[7]认为ATXN3基因突变患者出现帕金森综合征样表现可能与中低等长度的异常蛋白易于在锥体外系沉积有关。在发病早期,伴有锥体外系受累的SCA3很难与帕金森病鉴别,有阳性家族史且家系中有小脑体征的患者有助于诊断。此外,SCA3基因异常在家族性帕金森病患者中也可检测到,Park等[8]认为ATXN3基因突变是家族性帕金森病的原因之一。Wang等[9]对明确诊断的中国散发性及家族性帕金森病患者进行SCA3重复突变筛查显示,CAG重复序列数目异常可见于发病晚的中国人群(见于家族性帕金森病的3%及散发性帕金森病的0.8%),重复数为58~73,提示对有家族史的帕金森病患者可筛查该基因。
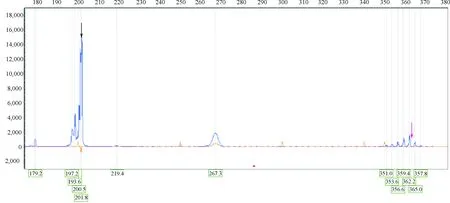
图 2 患者ATXN3基因检测结果(毛细管电泳法):ATXN3基因杂合突变,正常数目为7(黑色箭头所示),异常数目为61(红色箭头所示)
总之,以帕金森综合征为主要表现的SCA3病例较为少见,临床应予关注,对于具有家族史的帕金森综合征患者,需仔细查体,注意是否存在小脑体征和锥体束征,必要时进行ATXN3基因检测。
参考文献:
[1]Kim JS,Cho JW.Hereditary cerebellar ataxias: A Korean perspective[J].J Mov Disord,2015,8(2):67-75.
[2] Tang B,Liu C,Shen L,et al.Frequency of SCA1,SCA2,SCA3/MJD,SCA6,SCA7,and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds[J].Arch Neurol,2000,57:540-544.
[3] 王俊岭,吴允钦,雷立芳,等.中国汉族人群脊髓小脑性共济失调1、2、3、6、7、8、10、12、17亚型和齿状核红核苍白球路易体萎缩亚型多核苷酸正常重复次数范围研究[J].中华医学遗传学杂志,2010,27(5):501-505.
[4] 顾卫红,王国相,王康,等.脊髓小脑共济失调3型临床变异型特征及突变分析[J].中国现代神经疾病杂志,2008(2):134-138.
[5] Bettencourt C,Santos C,Coutinho P,et al.Parkinsonian phenotype in Machado-Joseph disease (MJD/SCA3): a two-case report[J].BMC Neurol,2011,11:131.
[6] Subramony SH,Hernandez D,Adam A,et al.Ethnic differences in the expression of neurodegenerative disease: Machado-Joseph disease in Africans and Caucasians[J].Mov Disord,2002,17(5):1068-1071.
[7] Maciel P1,Gaspar C,DeStefano AL,et al.Correlation between CAG repeat length and clinical features in Machado-Joseph disease[J].Am J Hum Genet,1995,57(1):54-61.
[8] Park H,Kim HJ,Jeon BS.Parkinsonism in spinocerebellar ataxia[J].Biomed Res Int,2015,Epub 2015.
[9] Wang JL,Xiao B,Cui XX,et al.Analysis of SCA2 and SCA3/MJD repeats in Parkinson’s disease in mainland China: genetic,clinical,and positron emission tomography findings[J].Mov Disord,2009,24(13):2007-2011.
