神经小胶质细胞在EAE脱髓鞘和髓鞘再生中的作用机制进展*
2018-04-20李国陵程晓东
叶 妮 李国陵 程晓东
(上海中医药大学附属岳阳中西医结合医院临床免疫研究所,上海 200437)
多发性硬化(Multiple sclerosis,MS)是最常见的中枢神经系统(Central nervous system,CNS)的自身免疫性疾病,其发病机制尚未明确。目前大多数学者认为其发病的关键是CD4+T细胞(主要是Th1和Th17)在外周自身髓鞘抗原的作用下异常激活,破坏血脑屏障(Blood brain barrier,BBB),在CNS内经抗原提呈细胞(Antigen presenting cell,APC)进一步激活,分泌大量炎性细胞因子攻击髓鞘,进而导致神经元死亡和轴突变性,引起一系列神经症状[1]。炎性细胞浸润和脱髓鞘是MS主要的病理特征,相关的病理研究多在其动物模型实验性自身免疫性脑脊髓膜炎(Experimental autoimmune encephalomyelitis,EAE)中开展。在CNS中,少突胶质细胞(Oligoden-drocyte,OL)是髓鞘形成细胞,神经小胶质细胞(Microglia,MG)是主要免疫效应细胞,星形胶质细胞(Astrocyte,AST)也具有免疫和分泌功能,三者与脱髓鞘和髓鞘再生过程密切相关。本文综述MG在EAE脱髓鞘和髓鞘再生中的直接作用机制,及其通过AST产生的间接作用机制及进展。
1 OL在EAE脱髓鞘和髓鞘再生中的作用
OL可形成致密、绝缘的多层膜性结构包绕轴突,即髓鞘,在促进神经元动作电位的跳跃式传递、营养轴突和维持神经可塑性中发挥重要作用。OL的损伤是脱髓鞘的直接原因,髓鞘再生也有赖于它的重新聚集。OL、AST和神经元均由神经干细胞发育分化而来,其中OL系可依据抗原表达、形态和功能特征可划分为5个渐变的发展阶段:前O2A祖细胞、O2A祖细胞、原OL、未成熟OL和OL。发育阶段早于OL的细胞统称为少突胶质前体细胞(Oligodendrocyte precursor cell,OPC)。促进OPC生存、迁移和增殖的细胞因子会抑制OPC的分化,如音猬因子(Shh)、血小板衍生生长因子AA (PDGF-AA)、成纤维细胞生长因子-2、趋化因子CXCL12等。也有胰岛素样生长因子-1、脑源性神经营养因子既可促进OPC增殖,也可促进其分化[2]。增殖分化后的OPC迁移至轴突附近,突起延伸并黏附至轴突,进一步发育为成熟的OL螺旋状包绕在轴突上,形成髓鞘。OL可以一个细胞同时发出多个板状突起包绕数条以至数十条的轴突,形成多条有髓神经纤维。
EAE是用髓鞘特异性抗原致敏易感动物,产生与MS相似的病理过程和临床表现的模型,已成为研究神经免疫学的主要工具。EAE的CNS内有大量炎性细胞浸润,其中直接损伤OL的细胞因子以Th1和MG分泌的肿瘤坏死因子-α(Tumor necrosis factor-α,TNF-α)和干扰素-γ(Interferon-γ,IFN-γ)为主导。TNF-α诱导OL凋亡的机制已经被很好地揭示,如促进凋亡诱导因子移位到细胞核、上调TNF相关凋亡诱导配体的诱骗受体3、4的表达、活化半胱天冬酶(Caspase)1、3、8等[3]。以往发现IFN-γ可通过促进FasR的表达引起OL的凋亡,最近新发现IFN-γ可激活胰内质网的未折叠蛋白应答,引起OL内质网的应激反应直接损伤OL[4]。值得一提的是,近来OL的损伤是CNS内炎症反应的原因还是结果引起了争议,这将MS的发病机制上升到更加复杂的高度。有研究使用转基因小鼠诱导性耗竭OL,发现OL丢失后出现了CD4+T细胞浸润到CNS的现象,随后由CNS内APC引起的炎症反应引发的是二次大规模脱髓鞘,即有可能是OL的丢失触发了髓鞘的自身免疫反应,引起MS[5]。尽管这个争议暂无定论,可以肯定的是CNS内的炎症反应极大地加速了OL的损伤。
OL是不能有丝分裂的细胞,丢失的OL必须由OPC分化补充,即髓鞘再生[6]。通常自发的髓鞘再生紧随脱髓鞘而发生,但是脱落的髓鞘碎片可激活补体,加重CNS内免疫应答,抑制髓鞘再生,也破坏残存的髓鞘[7]。高代谢活性、高能量需求、低抗氧化能力的OL对微环境的变化十分敏感,极易影响其成鞘能力。有研究表明,在炎症环境中,OPC比成熟的OL更敏感,更易受到损伤[8]。AST形成的胶质瘢痕作为物理屏障极大地影响了髓鞘再生,在EAE中已观察到OPC可以迁移到脱髓鞘区域,但是会被困在边缘,不能穿透到损伤内部[9]。因此随着EAE的病情进展,最终脱髓鞘部位的OPC减少,髓鞘再生是不完全的,甚至是失败的。
2 MG的功能特点
MG不仅具有结构支持、营养轴突和修剪突触等功能,作为CNS定居的巨噬细胞,也是CNS的主要免疫效应细胞,还具有免疫监视、提呈抗原和应答损伤的功能。依据巨噬细胞的分类,可将MG分为静息态M0型、促炎态M1型和抗炎态M2型。健康状态下,M0型MG呈分支状形态,发挥监视CNS微环境的作用,当CNS受到损伤时,MG可极化成M1或者M2型,变形为阿米巴样形态。M1型可分泌大量的促炎细胞因子及时杀死外周入侵的病原体,但同时也会损伤CNS。M2型可分泌抗炎细胞因子和神经营养因子,促进损伤修复,与M1型相配合,共同维持CNS内环境的稳定。然而,当CNS损伤反复出现时,M2型MG的抗炎作用逐渐减弱,不足以抵抗M1型诱发的炎症反应,导致疾病的持续发展。在体外,脂多糖(Lipopol-ysaccharide,LPS)、IFN-γ是强有力的M1型的诱导剂,M1型MG的标志物有诱导性一氧化氮合酶(inducible nitric oxide synthase,iNOS)、IFN-γ、TNF-α等。IL-4可有效地诱导M2型MG,以精氨酸酶-1(Arginase-1,Arg-1)、甘露糖受体(CD206)、IL-4、IL-10等为标志物,与M1型明显不同。需要注意的是,M1和M2型是MG活化后出现的两种极端的功能状态,还发现了中间态和待发态,总体MG是处于动态变化中[10]。
在EAE的发病初期,CNS内还没有外周来源的巨噬细胞的浸润,仅MG也可以发生免疫应答[11]。随着损伤的持续,BBB通透性的增加使得外周巨噬细胞进入CNS,与MG一起发挥免疫效应。由于激活的外周巨噬细胞和MG都呈现阿米巴样形态,难于分辨,很多研究在CNS内将其统称为巨噬细胞/MG。如已发现在EAE的早期阶段,是由M1型巨噬细胞/MG主导释放促炎细胞因子、活化效应T细胞,M2型巨噬细胞/MG主要是在损伤的高峰期及恢复期起抗炎作用,促进组织修复[12]。事实上,这两者也有一些不同之处。虽然这两类细胞的转录组很大程度上是共用的,但仍有至少600个转录产物是特异表达的,如MG的Tmem119、TREM-2等,外周巨噬细胞的CXCL13、CCR1等[13]。极化能力上,相比外周巨噬细胞,MG极化成M2型的能力受到限制。基因表达上,M1型巨噬细胞比M1型MG表达更多的抗原提呈标记物,如CD1A、1B和1C[14]。至于应答损伤,外周巨噬细胞只分布在CNS内的损伤区域,而MG也可在损伤周围分布。在损伤痊愈后,MG停留于CNS内,外周巨噬细胞则随血液循环离开CNS[11]。
3 M1型MG在EAE脱髓鞘中的作用
3.1提呈抗原 MG是固有免疫细胞,在CNS损伤时通过抗原提呈作用触发适应性免疫应答。早已知晓的是,为了有效地启动T细胞的活化,APC必须传递2个信号:一个来源于结合主要组织相容性复合体(Major histocompatibility complex,MHC)的抗原肽,另一个来源于共刺激分子。CNS内的APC主要指MG、AST、外周来源的巨噬细胞和树突状细胞(Dendritic cell,DC)。在提呈抗原的能力上,外周来源的DC活化初始T细胞更有效,而MG对浸润到CNS内的T细胞的再度活化起了关键性的作用[15]。静息时MG表达极低水平的MHCⅠ、MHCⅡ类分子和CD80、CD86等共刺激分子,但是在EAE的发病初期,MG表达的MHCⅡ类分子和共刺激分子明显上调,先于临床症状的出现,表明M1型MG可以有效地将抗原提呈给CD4+T细胞,通过表位扩展,引起新浸润的T细胞的激活[16]。此外,有研究发现MG表面MHCⅠ类分子的表达也出现了上升,表示MG也可以将抗原呈递给CD8+T细胞,可能是通过交叉呈递功能,即CD8+T细胞发挥的细胞毒溶解作用也参与了EAE的发病过程[17]。而随着EAE的病程进展,在恢复期MG表达的MHCⅡ、CD80、CD86明显降低,此时正是M2型MG占优势,表明主要是M1型MG而不是M2型MG发挥了抗原提呈作用[18]。T细胞再度活化后,可分泌促炎细胞因子、趋化因子等,激活更多的免疫细胞,迅速形成炎症级联,攻击OL。
3.2分泌促炎细胞因子 活化的T细胞分泌的促炎细胞因子,以及外周来源的免疫球蛋白、补体等,可以激活更多的MG,并诱导其向M1型极化。MG通过一系列免疫受体识别这些有害的刺激,如Toll样受体(Toll like receptor,TLR)、NOD样受体(NOD like receptor,NLR)和清道夫受体(Scavenger receptors,SR)[19]。此过程受到MAPK、JAK/STAT、PI3K/Akt等多条信号通路的调控,其中NF-κB作为共同的下游基因发挥了重要作用[20]。炎症环境下,IκB激酶复合体被激活,IκB磷酸化导致NF-κB核定位片段被暴露,迅速移入核内与特异性κB序列结合,NF-κB被激活,作为强效的炎性转录因子促使MG向M1型极化。此时MG和T细胞都可分泌促炎细胞因子,可相互活化,形成炎症级联。MG分泌的TNF-α、IFN-γ、IL-12可促进Th0向Th1极化,Th1进而释放IL-2、IL-12、IFN-γ、NF-κB,促进MG向M1极化;MG分泌的IL-1α、IL-1β、IL-6、IL-17、IL-23可促进Th0向Th17极化,Th17分泌的IL-1β、IL-6、IL-17、IL-22、IL-23、TNF-α、粒细胞-巨噬细胞集落刺激因子(Granulocyte macrophage colony stimulating factor,GM-CSF)又可加重炎症[16]。除了形成炎症级联,Th17分泌的IL-17、IL-22与BBB内皮细胞的IL-17R、IL-22R结合,下调紧密连接闭锁蛋白的表达,破坏BBB,从而促进免疫反应[21]。近来一些炎性小体,尤其是NLR家族的NLRP3被认为在MS发病中起了关键性的作用。有研究发现GM-CSF可以激活NLRP3,促进IL-1β和IL-18的成熟和分泌,而IL-18可增强Th0向Th1和Th17的极化[22]。由此我们总结,虽然直接损伤OL的细胞因子以TNF-α和IFN-γ为主导,但是M1型MG和T细胞形成的炎症级联可多途径地促进这两种细胞因子的分泌,造成脱髓鞘。
3.3诱发氧化应激 炎症环境下,MG的能量代谢方式出现了明显的变化:常氧时,细胞的能量代谢是在葡萄糖转化为丙酮酸之后,通过氧化磷酸化进入三羧酸循环;而在MG向M1型极化时,丙酮酸主要通过无氧糖酵解进行代谢,伴随着葡萄糖消耗增加和乳酸聚集。糖酵解使MG快速产生大量的活性氧物质(Reactive oxygen species,ROS)、活性氮物质(Reactive nitrogen species,RNS)以及NADPH氧化酶,通过增加内部环境的酸度和剥夺微生物的铁抑制新陈代谢以发挥杀伤异物的功能[23]。同时,炎症激活了NF-κB、STAT等信号,低氧环境促使了缺氧诱导因子-1(HIF-1)的产生,由于iNOS的启动子区域含有多个炎症和低氧相关转录因子的结合位点,iNOS可以大量表达[24]。iNOS使用NADPH提供的电子,通过氧化L-精氨酸,大量合成一氧化氮(Nitric oxide,NO)。NO本身作为第二信使和神经递质,在血管舒张、吞噬、抗肿瘤、学习和记忆方面起了重要作用,而大量的NO可作为氧自由基损伤DNA,使DNA突变,作为信号分子引起p53和TNF介导的细胞凋亡,直接损伤OL和OPC[25]。更重要的是,NO可与ROS中超氧阴离子结合生成过氧亚硝酸盐(Peroxynitrite,ONOO-),ONOO-是最活跃的RNS,作为强氧化剂破坏机体正常的抗氧化机制,破坏正常的代谢和膜功能,硝基化正常蛋白质的酪氨酸以及强烈抑制线粒体的氧化呼吸,有广泛的细胞毒作用[26]。大量的ONOO-还可下调兴奋性氨基酸转运蛋白的表达,使谷氨酸积累于细胞外,引起兴奋毒性损伤[27]。由此我们总结,由于MG能量代谢方式的改变引起一系列氧化应激产物的聚集,可以全面损伤CNS内的细胞,尤其是环境敏感的OL和OPC,也是造成脱髓鞘的重要原因。
3.4分泌趋化因子 生理状态下,MG并不表达炎性趋化因子及其受体,但是在炎症环境下,T细胞和MG都高表达一些趋化因子,成为EAE的另一大特点。总体上,M1型MG高表达CCL2-CCL5、CCL8、CCL12、CCL19、CCL21、CCL22、CXCL1、CXCL3、CXCL8-CXCL13,及其受体CCR3、CXCR1、CXCR3等,可趋化MG、AST、T细胞、B细胞、DC等向CNS的病灶部位聚集,促进局部炎症[28]。其中,TNF-α、IL-1β、IFN-γ可以诱导MG产生CCL2,CCL2被发现不仅能够招募CCR2+CD11b+ly6Chi单个核细胞到CNS中,还破坏了BBB完整性[29]。虽然AST和内皮细胞也可产生CCL2,并且在EAE的不同阶段发挥了作用,但是特异性消除AST和内皮细胞来源的CCL2后没有抑制EAE的发病,所以MG来源的CCL2在诱导EAE中发挥了重要作用[30]。
3.5促进AST的脱髓鞘作用 虽然AST是CNS内提呈抗原能力最弱的APC,但是由于其在CNS内庞大的数量,它的免疫和分泌功能也必须考虑到。人类和鼠AST只构成性的表达一种TLR是TLR3,可被IFN-γ、IL-1β触发上调,即炎症环境可以激活AST[31]。作为BBB的重要组成成分,活化的AST丢失了在小血管周围的终足,导致BBB通透性增加,是EAE发病的早期事件。EAE时,AST也可分泌促炎细胞因子与M1型MG、T细胞相互活化,促进炎症级联,如IL-1β、IL-12、IL-23、IL-17、NF-κB[32]。此时AST表达的谷氨酸转运体、谷氨酰胺合成酶与谷氨酸脱氢酶降低,表示细胞外谷氨酸的积累,引起OL的兴奋毒性损伤[33]。此外,AST分泌的一些趋化因子,如CX32和CX47在CNS脱髓鞘区域严重降低,表示OL-OL、OL-AST之间的缝隙连接丢失,导致脱髓鞘[34]。对于髓鞘再生,除了前述胶质瘢痕的存在阻碍了OPC进入脱髓鞘区域内部,活化的AST还分泌了一些细胞外基质成分,如硫酸软骨素蛋白多糖(CSPG)、高分子的透明质酸(HA),可抑制OPC的生长、分化和黏附,抑制髓鞘再生[35]。虽然我们并不能确定是否是MG最先激活了AST,但是M1型MG产生的炎症级联必然极大地促进了AST的脱髓鞘作用。
4 M2型MG在EAE髓鞘再生中的作用
4.1结合负性共刺激信号 由于MG是CNS内固有的APC,它不仅可以结合CD80、CD86等正性共刺激信号,也可以结合负性共刺激信号,抑制T细胞应答,有利于髓鞘再生,目前相关的研究集中在程序性死亡受体-1(Programmed death-1,PD-1)。PD-1在活化的T细胞表面高度表达,与其配体PD-L1或者PD-L2结合后,通过磷酸化胞质区的免疫受体酪氨酸转化基序(ITSM)传导抑制性信号[36]。体外诱导的M1型MG高表达CD86和PD-L1,低表达PD-L2;M2型MG高表达PD-L2,中度表达PD-L1[37]。在EAE的研究中发现,随着病程的进展,CNS中浸润的CD4+T细胞逐渐表达PD-1,在高峰期达到峰值,随后下降,与MG表达的PD-L1趋势一致,而MG表达的PD-L2呈平稳状态。这表明M1和M2型MG主要通过PD-L1与CD4+T细胞表面的PD-1结合而发挥免疫抑制作用,与EAE的自限性相关,M2型MG表达的PD-L2对T细胞的抑制作用还需要深入的研究[18]。通过PD-1敲除的小鼠发现,PD-1是通过降低STAT-1的磷酸化水平、抑制NF-κB信号而抑制MG向M1型极化,增加STAT-6的磷酸化水平而促进其向M2型极化[38]。最近有研究发现CD40及其配体CD40L在M1型MG表面高表达,其可与Th1和Th17之间形成负反馈循环机制,或可成为一个抑制过度炎症反应的免疫检查点[39]。然而M2型MG并非静息态MG,CD40和CD40L在M2型MG中的表达情况及意义目前未见报道。
4.2分泌抗炎细胞因子 在EAE的高峰期及缓解期,CNS中浸润的CD4+T细胞中Th2逐渐取代Th1,释放抗炎细胞因子,IL-4、IL-10、IL-13等。这些细胞因子可以抑制M1型MG的活性,诱导MG向M2型极化。M2型MG进一步分泌IL-4、IL-10、IL-13、转化生长因子-β(Transforming growth factor-β,TGF-β)、过氧化物酶体增殖物激活受体γ(Peroxisome proliferator activated receptor γ,PPARγ)等,抑制CNS内炎症应答,也可诱导Th2和调节性T细胞(regulatory T cell,Treg)的产生[40]。在向M2型的极化过程中,STAT-6通路起了关键作用,IL-4、IL-13分别与MG表面的IL-4R、IL-13R结合,IL-10与MG表面的IL-10R1/2结合都可激活STAT-6,释放抗炎细胞因子[41]。有趣的是,一些促炎细胞因子,如TNF-α、IFN-γ也被证明具有保护髓鞘的功能。有研究发现在EAE中阻断了TNF-α后,虽然降低了发病率、延迟了发病时间,但是增加了脾中Th1和Th17应答,小鼠CNS内OPC和OL数量减少,髓鞘再生被明显的延迟[42]。究其原因,是因为TNF-α的受体有TNFR1和TNFR2之别,TNFR1在CNS中主要诱导细胞凋亡,而TNFR2是促进OL分化的重要分子[43]。有研究发现用药物治疗EAE后,随着M2型MG的产生,IL-10的分泌增加,但是同时IFN-γ不降反增,表明IFN-γ和IL-10一样有保护髓鞘作用[44]。早年发现IFN-γ对髓鞘的作用依赖其浓度变化,体外低浓度的IFN-γ刺激后的MG可以促进OL的形成,而高浓度时抑制OPC分化,并且这种抑制作用可被IL-4消除[45]。近来有研究发现它还可以抑制髓鞘脂质过氧化产物的产生,促进髓鞘碎片的清除,丰富了IFN-γ的髓鞘保护机制[46]。值得注意的是,TGF-β作为抗炎细胞因子之一,在高浓度时可诱导Treg的产生,在低浓度时也可诱导初始T细胞向Th17分化,促进EAE的发展[47]。
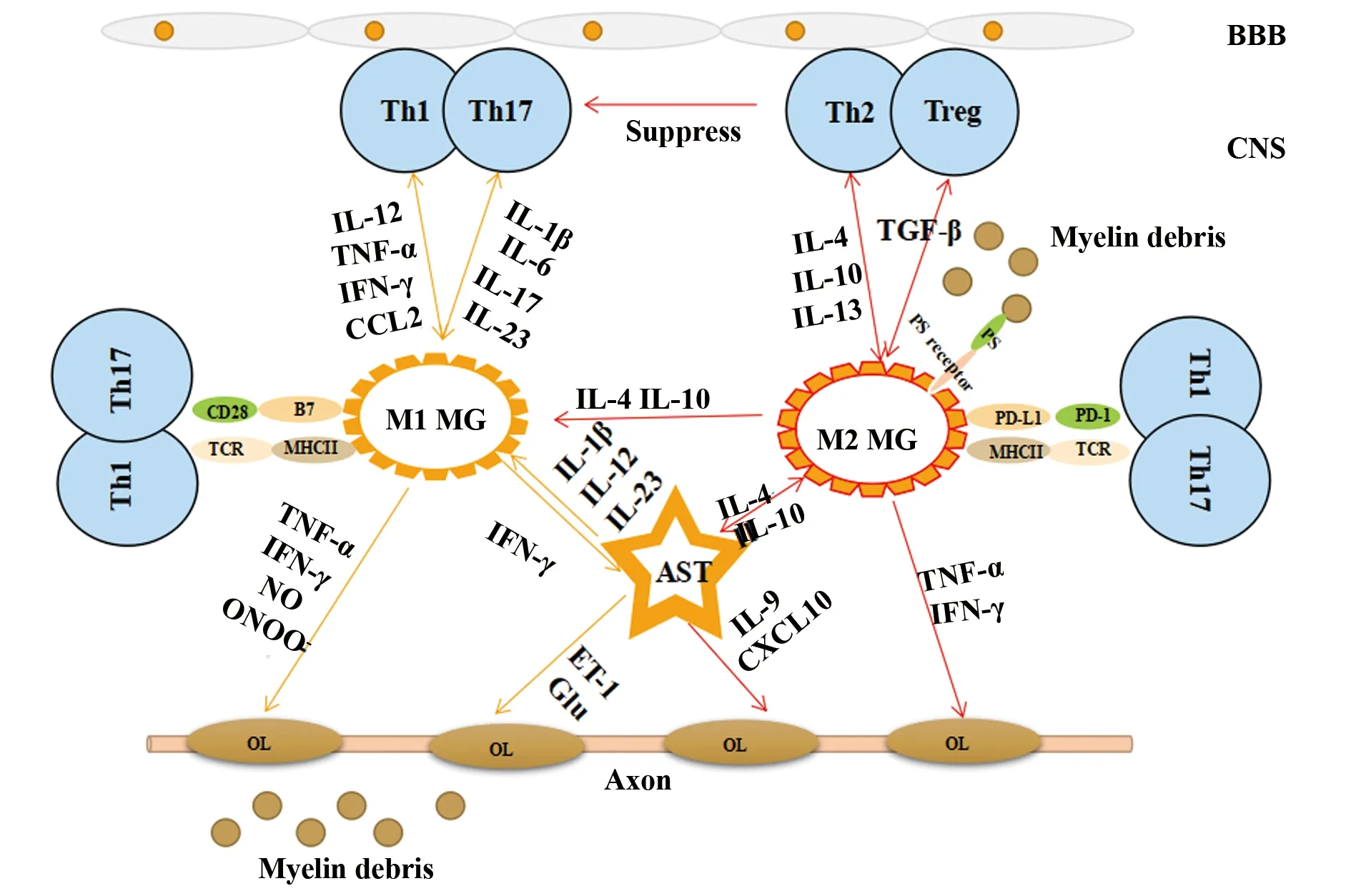
图1 神经小胶质细胞在EAE脱髓鞘和髓鞘再生的关键作用机制Fig.1 Key mechanisms of microglial function on demye-lination and remyelination in EAE modelNote:Yellow arrows mean demyelination and red arrows mean remyelination.
4.3吞噬髓鞘碎片 吞噬作用是巨噬细胞的一项基本生物学功能,M2型MG发挥的吞噬髓鞘碎片作用是促进髓鞘再生的重要途径。与M1型相比,M2型MG的葡萄糖消耗量显著降低,主要通过氧化磷酸化进行能量代谢,与未极化的MG相似[48]。此时精氨酸代谢为鸟氨酸和多胺,通过调节能量需求和膜流动性有助于吞噬作用[49]。吞噬髓鞘碎片依赖于一系列胞膜上的磷脂酰丝氨酸(Phosphati-dylserine,PS)受体,如Mer-TK、TREM-2、SIRP-α。EAE中,脱落的OL胞膜上的PS外翻,M2型MG表面的PS受体与之结合,从而成功地吞噬髓鞘碎片,抑制免疫级联的发生,为髓鞘再生提供空间[50]。尽管M1型MG也可表达相似的PS受体,但其吞噬髓鞘碎片能力相对较弱。在CNS中,由于瓦勒变性,髓鞘碎片的清除是缓慢和不完全的,相比外周巨噬细胞,MG有效清除碎片的能力大幅降低[51]。MG的吞噬能力也非越强越有利,有研究发现其过度的吞噬还可以引起呼吸爆发,造成氧化应激[52]。
4.4促进AST的髓鞘再生作用 在EAE的晚期阶段,由于Th2和M2型MG的增加,激发纤维型AST增生,在髓鞘再生时表现出一定的促进作用。先前被证明AST来源的CXCL10可以抑制OPC的分化,但是也有研究表明AST有招募MG到CNS损伤区域的功能,进而吞噬髓鞘碎片,这个过程正是由AST分泌的CXCL10调节的[53]。AST也可以产生IL-4、IL-10、TGF-β等抗炎细胞因子抑制MG的活化和MHCⅡ类分子的表达[54]。EAE时,AST高表达IL-9,IL-9与其他促炎细胞因子联合,会抑制OPC增殖和分化,但与IFN-γ联合能促使OPC增殖和分化[55]。此外,AST也被发现有吞噬凋亡细胞的能力,依赖于AST附近的微环境,其吞噬能力不如MG强大[56]。由AST形成致密的胶质瘢痕,在某种程度上可以保护周围细胞免受胞外钾、谷氨酸、ROS、RNS等水平升高导致的继发变性,有利于髓鞘再生。
5 总结与展望
综上所述,M1型MG可通过提呈抗原、分泌促炎细胞因子、诱发氧化应激、分泌趋化因子的直接作用引发脱髓鞘,还可通过AST间接地损伤髓鞘,在EAE发病的早期起到了推动作用。而在EAE的高峰期和恢复期,M2型MG占优势,通过结合负性共刺激信号、分泌抗炎细胞因子、吞噬髓鞘碎片的途径抑制M1型MG的作用,也可通过AST间接地促进髓鞘再生。在未来,基于M1和M2型MG的功能特性,在MS的发病早期调节M1/M2型MG的比例,使M2型MG占优势有望成为治疗MS的有效途径。比如通过抑制NF-κB信号、促进STAT-6通路、抑制M1型MG的氧化应激水平、适度地增强M2吞噬髓鞘碎片的能力等。但是大量的增加M2型MG,抑制M1型MG的产生,是否会因CNS内防御能力的下降而诱发新的问题,还需要深入探索。
参考文献:
[1] Rangachari M,Kerfoot SM,Arbour N,etal.Editorial:lymphocytes in MS and EAE:more than just a CD4+world[J].Front Immunol,2017,8:133.
[2] Mitew S,Hay CM,Peckham H,etal.Mechanisms regulating the development of oligodendrocytes and central nervous system myelin[J].Neuroscience,2014,276(2014):29-47.
[3] Chen KB,Uchida K,Nakajima H,etal.Tumor necrosis factor-α antagonist reduces apoptosis of neurons and oligodendroglia in rat spinal cord injury[J].Spine (Phila Pa 1976),2011,36(17):1350-1358.
[4] Lin Y,Huang G,Jamison S,etal.PERK activation preserves the viability and function of remyelinating oligodendrocytes in immune-mediated demyelinating diseases[J].Am J Pathol,2014,184(2):507-519.
[5] Traka M,Podojil JR,McCarthy DP,etal.Oligodendrocyte death results in immune-mediated CNS demyelination[J].Nat Neurosci,2016,19(1):65-74.
[6] Dietz KC,Polanco JJ,Pol SU,etal.Targeting human oligodendrocyte progenitors for myelin repair[J].Exp Neurol,2016,283(Pt B):489-500.
[7] Kopper TJ,Gensel JC.Myelin as an inflammatory mediator:myelin interactions with complement,macrophages,and microglia in spinal cord injury[J].J Neurosci Res,2017,doi:10.1002/jnr.24114.
[8] Cui QL,Kuhlmann T,Miron VE,etal.Oligodendrocyte progenitor cell susceptibility to injury in multiple sclerosis[J].Am J Pathol,2013,183(2):516-525.
[9] Bannerman P,Hahn A,Soulika A,etal.Astrogliosis in EAE spinal cord:derivation from radial glia,and relationships to oligodendroglia[J].Glia,2007,55(1):57-64.
[10] Tang Y,Le W.Differential roles of M1 and M2 microglia in neurodegenerative diseases[J].Mol Neurobiol,2016,53(2):1181-1194.
[11] Yamasaki R,Lu H,Butovsky O,etal.Differential roles of microglia and monocytes in the inflamed central nervous system[J].J Exp Med,2014,211(8):1533-1549.
[12] Rawji KS,Yong VW.The benefits and detriments of macrophages/microglia in models of multiple sclerosis[J].Clin Dev Immunol,2013,2013:948976.
[13] Hickman SE,Kingery ND,Ohsumi TK,etal.The microglial sensome revealed by direct RNA sequencing[J].Nat Neurosci,2013,16(12):1896-1905.
[14] Durafourt BA,Moore CS,Zammit DA,etal.Comparison of polarization properties of human adult microglia and blood-derived macrophages[J].Glia,2012,60(5):717-727.
[15] Chastain EM,Duncan DS,Rodgers JM,etal.The role of antigen presenting cells in multiple sclerosis[J].Biochim Biophys Acta,2011,1812(2):265-274.
[16] Strachan-Whaley M,Rivest S,Yong VW.Interactions between microglia and T cells in multiple sclerosis pathobiology[J].J Interferon Cytokine Res,2014,34(8):615-622.
[17] Goverman J.Autoimmune T cells responses in the central nervous system[J].Nat Rev Immunol,2009,9(6):393-407.
[18] Hu J,He H,Yang Z,etal.Programmed death ligand-1 on microglia regulates Th1 differentiation via nitric oxide in experimental autoimmune encephalomyelitis[J].Neurosci Bull,2016,32(1):70-82.
[19] Ransohoff RM,Browm MA.Innate immunity in the central nervous system[J].J Clin Invest,2012,122(4):1164-1171.
[20] Merson TD,Binder MD,Kilpatrick TJ.Role of cytokines as mediators and regulators of microglia activity in inflammatory demyelination of the CNS[J].Necro Mol Med,2010,12(2):99-132.
[21] Huppert J,Closhen D,Croxford A,etal.Cellular mechanisms of IL-17 induced blood brain barrier disruption[J].FASEB J,2010,24(4):1023-1034.
[22] Barclay W,Shinohara ML.Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE)[J].Brain Pathol,2017,27(2):213-219.
[23] Orihuela R,McPherson CA,Harry GJ.Microglial M1/M2 polarization and metabolic states[J].Br J Pharmacol,2016,173(4):649-665.
[24] Robinson MA,Baumgardner JE,Otto CM.Oxygen-dependent regulation of nitric oxide production by inducible nitric oxide synthase[J].Free Radic Biol Med,2011,51(11):1952-1965.
[25] Nadjafi S,Ebrahimi SA,Rahbar-Roshandel N.Effect of berberine on nitric oxide production during oxygen-glucose deprivation/reperfusion in OLN-93 oligodendrocytes[J].Pak J Biol Sci,2014,17(11):1185-1189.
[26] Li L,Zhao D,Li J.Alteration of apoptotic protease activating factor 1 expression and possible role in ONOO(-)-induced apoptosis in human cerebral vascular smooth muscle cells[J].Int J Clin Exp Med,2015,8(10):19739-19745.
[27] Vercellino M,Masera S,Lorenzatti M,etal.Demyelination,inflammation,and neurodegeneration in multiple sclerosis deep gray matter[J].J Neuropathol Exp Neurol,2009,68(5):489-502.
[28] Ramesh G,MacLean AG,Philipp MT.Cytokines and chemokines at the crossroads of neuroinflammation,neurodegeneration,and neuropathic pain[J].Mediators Inflamm,2013,2013:480739.
[29] Semple BD,Kossmann T,Morganti-Kossmann MC.Role of chemokines in CNS health and pathology:a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks[J].J Cereb Blood Flow Metab,2010,30(3):459-473.
[30] Paul D,Ge S,Lemire Y,etal.Cell-selective knockout and 3D confocal image analysis reveals separate roles for astrocyte-and endothelial-derived CCL2 in neuroinflammation[J].J Neuroinflammation,2014,11:10.
[31] Sal mina AB,Komleva YK,Lopatina OL,etal.Astroglial control of neuroinflammation:TLR3-mediated dsRNA-sensing pathways are in the focus[J].Rev Neurosci,2015,26(2):143-159.
[32] Chanaday NL,Roth GA.Microglia and astrocyte activation in the frontal cortex of rats with experimental autoimmune encephalomyelitis[J].Neuroscience,2016,314:160-169.
[33] Murugan M,Ling EA,Kaur C.Dysregulated glutamate uptake by astrocytes causes oligodendroglia death in hypoxic perventricular white matter damage[J].Mol Cell Neurosci,2013,56:342-354.
[34] Markoullis K,Sargiannidou I,Gardner C,etal.Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis.[J] Glia,2012,60(7):1053-1066.
[35] Stoffels JM,Hoekstra D,Franklin RJ,etal.The EIIIA domain from astrocyte-derived fibronectin mediates proliferation of oligodendrocyte progenitor cells following CNS demyelination[J].Glia,2015,63(2):242-256.
[36] Dai S,Jia R,Zhang X,etal.The PD-1/PD-Ls pathway and autoimmune diseases[J].Cell Immunol,2014,290(1):72-79.
[37] Zhang XM,Lund H,Mia S,etal.Adoptive transfer of cytokine-induced immunomodulatory adult microglia attenuates experimental autoimmune encephalomyelitis in DBA/1 mice[J].Glia,2014,62(5):804-817.
[38] Yao A,Liu F,Chen K,etal.Programmed death 1 deficiency induces the polarization of macrophages/microglia to the M1 phenotype after spinal cord injury in mice[J].Neurotherapeutics,2014,11:636-650.
[39] Gaikwad S,Patel D,Agrawal-Rajput R,etal.CD40 negatively regulates ATP-TLR4-activated inflammasome in microglia[J].Cell Mol Neurobiol,2017,37(2):351-359.
[40] Correale J.The role of microglial activation in disease progression[J].Mult Scler,2014,20(10):1288-1295.
[41] Yin L,Chen Y,Qu Z,etal.Involvement of JAK/STAT signaling in the effect of cornel iridoid glycoside on experimental autoimmune encephalomyelitis amelioration in rats[J].J Neuroimmunol,2014,274(1-2):28-37.
[42] Batoulis H,Recks MS,Holland FO,etal.Blockade of tumour necrosis factor-α in experimental autoimmune encephalomyelitis reveals differential effects on the antigen-specific immune response and central nervous system histopathology[J].Clin Exp Immunol,2014,175(1):41-48.
[43] Madsen PM,Motti D,Karmally S,etal.Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination[J].J Neurosci,2016,36(18):5128-5143
[44] Benedek G,Zhang J,Bodhankar S,etal.Estrogen induces multiple regulatory B cell subtypes and promotes M2 microglia and neuroprotection during experimental autoimmune encephalomyelitis[J].J Neuroimmunol,2016,293:45-53.
[45] Butovsky O,Ziv Y,Schwartz A,etal.Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells[J].Mol Cell Neurosci,2006,31(1):149-160.
[46] Sosa RA,Murphey C,Robinson RR,etal.IFN-γ ameliorates autoimmune encephalomyelitis by limiting myelin lipid peroxidation[J].Proc Natl Acad Sci U S A,2015,112(36):E5038-5047.
[47] Ghaedi M,Namdari H,Rahimzadeh P,etal.Different doses of transforming growth factor-β on in vitro differentiation of human naïve CD4+T cells to T helper 17[J].Iran J Allergy Asthma Immuno,2015,14(6):633-637.
[48] Blagih J,Jones RG.Polarizing macrophages through reprogram ming of glucose metabolism[J].Cell Metab,2012,15(6):793-795.
[49] Mills CD.M1 and M2 macrophages:oracles of health and disease[J].Crit Rev Immunol,2012,32(6):463-488.
[50] Sierra A,Abiega O,Shahraz A,etal.Janus-faced microglia:beneficial and detrimental consequences of microglial phagocytosis[J].Front Cell Neurosci,2013,7:6.
[51] Shinjo R,Imagama S,Ito Z,etal.Keratan sulfate expression is associated with activation of a subpopulation of microglia/macrophages in Wallerian degeneration[J].Neurosci Lett,2014,579:80-85.
[52] Claude J,Linnartz-Gerlach B,Kudin AP,etal.Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst[J].J Neurosci,2013,33(46):18270-18276.
[53] Skripuletz T,Hackstette D,Bauer K,etal.Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination[J].Brain,2013,136(Pt 1):147-167.
[54] Arbo BD,Bennetti F,Ribeiro MF.Astrocytes as a target for neuroprotection:modulation by progesterone and dehydroepiand-rosterone[J].Prog Neurobiol,2016,144:27-47.
[55] Ding X,Cao F,Cui L,etal.IL-9 signaling affects central nervous system resident cells during inflammatory stimuli[J].Exp Mol Pathol,2015,99(3):570-574.
[56] Ponath G,Ramanan S,Mubarak M,etal.Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology[J].Brain,2017,140(2):399-413.
