气相色谱-燃烧-稳定同位素比值质谱测定葡萄酒中甘油δ13C的可行性探讨
2018-04-04王道兵钟其顶李国辉岳红卫
王道兵 ,钟其顶 ,李国辉 ,岳红卫
(1.中国食品发酵工业研究院有限公司,北京 100015; 2.全国食品发酵标准化中心,北京 100015)
甘油是葡萄酒中干浸出物的主要化合物,主要产生于葡萄酒发酵生产过程。虽然其对葡萄酒的香气特征没有直接贡献,但却赋予葡萄酒酒体丰满的特征——不仅可以增加葡萄酒的甜度,而且使葡萄酒口感更厚重圆润[1],因此一些欧洲国家也将甘油作为葡萄酒评级的依据[2]。因葡萄质量不好或者发酵过程的问题会导致生产出来的葡萄酒口感较差,虽然各国标准与法规禁止人为加入甘油,但一些不法酒商仍会添加甘油来改善葡萄酒的口感,更有甚者直接用工业甘油和三精一水化学原料生产“葡萄酒”。
稳定同位素技术在葡萄酒掺假检测中具有重要应用[3],利用稳定碳同位素的自然分馏特征检测葡萄酒中非法添加的甘油组分,对维护葡萄酒市场公平竞争、保护消费者权益、打击葡萄酒造假具有重要意义。自1997年以来,关于甘油碳同位素分析的报道有很多[4-8],其中,Weber等[4]和Rossmann等[5]首先分析了不同产品中甘油的碳同位素分布特征,Fronza等[6-7]研究了动物脂肪转化的甘油同位素特点,陈珊珊等[8]测定了植物油中甘油碳同位素组成,然而上述方法仅限于分析甘油含量较高的样品——样品中甘油提纯后再测定。随着在线分析技术的发展,气相色谱-燃烧-稳定同位素比值质谱(GC-C-IRMS)在有机物碳同位素分析中的研究与应用越来越广泛,其分析精度也可优于0.3‰[9-10]。2004年,Calderone报道了直接进样模式联用GC-CIRMS测定葡萄酒中甘油碳同位素比值的方法[11],该方法也被国际葡萄与葡萄酒组织(OIV)采纳为标准方法(Resolution OIV/OENO 343/2010),然而我们在应用OIV方法时发现其存在一些问题:葡萄酒样品用乙醇稀释4倍后取0.3 μL进样(分流比120∶1,进样口温度270℃)测定,却未能监测到甘油的色谱峰,为此,我们针对样品的不同属性进行了方法验证。
GC-C-IRMS适于气体或较低沸点物质的碳氮同位素特征分析,然而常压下甘油的沸点是290℃,在205℃或稍高温度时会随受热时间而有不同程度的聚合和分解,在此过程中必然会发生同位素动力学分馏[12-13]。根据稳定同位素分析“IT”原则[14-15],测定样品甘油δ13C时只需确保和标准品中的甘油具有相同的气化特征而无需完全气化,但是考虑到葡萄酒中甘油含量仅为4~11 g/L,而IRMS精确分析需要约200 ng C[16],因此,探讨色谱条件对甘油气化和碳同位素分馏的影响对准确测定葡萄酒甘油δ13CVPDB值具有重要意义。本研究针对上述因素对测定结果的影响展开了探讨,以期为我国葡萄酒中甘油的稳定同位素分布特征调查和真实性应用研究选择合适的技术方法提供数据支持。
1 材料与方法
1.1 材料、试剂及仪器
葡萄酒:市售。
仪器设备:Delta V Advantage稳定同位素比值质谱仪,配备Ultra Trace GC气相色谱、IsoLink接口和Triplus自动进样器,以及Flash 2000元素分析仪,以上仪器均购自Thermo Fisher公司。超纯水由Milipore公司的Milli-Q系统制备。氦气(纯度为99.999%)和二氧化碳气体(纯度99.99%)均购于北京北温气体制造厂。HP-INNOwax气相色谱柱(30 m×0.25 mm×0.25µm)购于安捷伦科技(中国)有限公司。十万分之一电子天平购于瑞士Mettler-Toledo公司。
试剂:乙醇(色谱纯),购于国药集团化学试剂有限公司;甘油(分析纯),编号为WS-1、WS-2和WS-3,购于国药集团化学试剂有限公司。
IAEA-CH-6(国际原子能机构),δ13CV-PDB=-10.45‰±0.2 ‰;IAEA-601(国际原子能机构),δ13CV-PDB=-27.77‰±0.04‰。
1.2 仪器条件
1.2.1 GC条件
进样量1 μL,进样口温度240~270℃,载气为氦气,流速1.2 mL/min,分离比10∶1。程序升温条件为:初温120℃,升温速度为25℃/min。
1.2.2 甘油转化条件
燃烧转化装置(IsoLink)中配备陶瓷(Al2O3)氧化管(填料为CuO、NiO 和Pt),工作温度为1000 ℃[12-13]。
1.2.3 EA条件[15]
氧化管温度980℃,还原管温度680℃,柱温60℃,氦气流速100 mL/min,氧气充入时长为3 s(流速250 mL/min);称取0.1 mg有机物于锡杯中,包样后测定。
1.2.4 IRMS条件
离子源电压2.97 kV,真空度1.8×10-6mbar,轰击电压120.8 eV。
1.3 数据处理
Thermo isodat version 3.0软件用于数据采集和碳同位素值的计算(注:δ13C值仅是实际测定结果,未经过标准品校正。校正后的结果记为δ13CVPDB)。
2 结果与讨论
受同位素热力学分馏作用的影响,物质在两种或两种以上物质(物相)之间的分配时具有不同的同位素比值,而分馏系数取决于物质分配时的条件,如温度、溶剂等[17]。GC-C-IRMS系统中存在两个分配节点:进样口和燃烧管/裂解管接口,本研究中,我们重点针对上述两个节点中有样品损失的情况而重点分析温度、溶剂对甘油碳同位素分馏的影响。
2.1 甘油工作标准
由于国内外均缺乏甘油碳同位素参考物质,因此我们选择国际碳同位素参考物质标定的方法得到甘油工作标准物质:用IAEA-CH-6和IAEA-601反标定EA-IRMS中参考气CO2的δ13CVPDB[18],然后多次测定纯甘油的稳定碳同位素比值并取其平均值,得到3个甘油WS-1、WS-2和WS-3的 δ13CVPDB值分别为-30.98‰±0.05‰、-27.95‰±0.04‰和-25.08‰±0.04‰。由此可见,上述3个纯甘油具有不同的碳同位素特征且均匀稳定,因而可以作为实验室工作标准用于方法验证和同位素分馏特征研究。
2.2 进样口温度对葡萄酒甘油测定的影响
Schmitt等[19]的研究表明,进样口参数设置会影响气体物质分析时的同位素分馏特征,而为了保持良好的分离度和峰形,我们一般采用分流进样的模式,这种分流造成的样品损失必然也会带来同位素分馏。研究进样口条件对GC-C-IRMS测定甘油δ13C的影响,保持色谱柱温度240℃,设定IsoLink的反吹阀(Backflush valve)在0~300 s处于打开状态以防止乙醇进入反应管。以甘油乙醇溶液(WS-1、WS-2、WS-3)和葡萄酒(Wine-01)为研究对象,分别在进样口温度200℃、240℃、270℃和300℃时进样,每个试样进样3次,测试序列中以270℃条件下的测定结果为基准进行结果初校正。甘油经GC-C-IRMS转化的CO2的信号强度(m/z44)见图1,δ13C结果见表1。

图1 不同进样口温度时甘油转化的CO2的信号强度(m/z44)
从图1可看出,信号强度(m/z44)因进样口温度不同而有差异:小于240℃时,信号强度(m/z44)随温度升高而增强;大于270℃时,信号强度(m/z44)与温度呈反相关关系;乙醇为溶剂的甘油样品在240℃和270℃时几乎是一样的,但葡萄酒中甘油的信号强度(m/z44)在270℃条件下达到最高值。这与甘油在气相色谱进样口的气化特性有关,低于240℃时有相当一部分甘油未气化而未能进入色谱柱;高于270℃时,可能会导致甘油分解,也可能是高温条件下的乙醇、水等其他低沸点物质的蒸汽分压太大,导致甘油的气化占比变小,而进入色谱柱的比例也随之降低的缘故。
表1数据表明,试样在4个温度条件下δ13C值的标准偏差均小于0.3‰,尤其是WS-1,其不同温度下测定值的标准偏差均优于0.20‰,考虑到进样口温度270℃时,Wine-01的甘油具有更高的信号强度(m/z44),后续研究中均固定进样口温度为270℃。

表1 甘油在不同进样温度下的δ13C测定结果 (‰)
对比各样品的甘油δ13C实测值可以看出,不同温度条件下测定结果均不相同,并且呈现出随进样口温度升高而δ13C值下降的趋势,并且甘油中13C/12C比值越大,测定结果变化的程度也越大,这表明GC-C-IRMS测定甘油碳同位素时,在进样口处出现了碳同位素分馏,温度越高时,质量数大的甘油分子越容易进入色谱柱,这符合瑞利分馏的相关特征[20]。
由2.1可知,3个甘油工作标准物质的δ13CVPDB差值(Δ13C)最大为5.90‰(WS-3与WS-1),然而GC-C-IRMS测定结果(δ13C值)的差值却不是5.90‰,而是在6.43‰~7.34‰范围内变化,显然,EA-IRMS与GC-C-IRMS在分析样品时具有不同的同位素分馏特征,若以WS-1为基准进行数据校正,WS-3的校正结果与EA-IRMS标定值也会存在偏差,偏差的大小与进样口温度密切相关。结合分馏系数(ɑ)因温度而变的特点,说明只依靠工作参考气CO2或单点校正法计算样品的δ13C值是不充分的,而应该按照“PIT”原则,并且依据Stephen等[22]建立的两点校正模型来进行数据校正:以进样口270℃下的测定结果为例,3个标样的测定值与给定值之间的线性拟合程度R2=1,这说明该分析条件(进样口270℃、色谱柱240℃恒温、分流进样10∶1)下甘油碳同位素分馏特征是线性的、分馏系数是固定的。采用两点校正法,任取2个标样的测定值和给定值建立回归曲线,如WS-1和WS-2之间的线性拟合公式为δ13CVPDB=0.8832*δ13C-6.5792,由此计算得出WS-2的δ13CVPDB值为-27.93‰,与给定值-27.95‰十分接近。
2.3 色谱柱温度对甘油测定的影响
如上文所述,分流进样模式下进样口温度影响样品的气化特征进而影响甘油产生的CO2的信号强度,同样地,GC-C-IRMS体系中毛细色谱柱并非直接连在检测器上,色谱柱流出的物质约有90%的物质转入燃烧管中(其余10%经裂解管排出)。甘油在该节点处并未100%进入燃烧管,因此碳同位素分馏在该处同样存在。为验证色谱柱温度对甘油δ13C测定的影响,在进样口温度270℃、载气流速1.2 mL/min的情况下,设定程序升温的初始温度为80℃,保持1 min,然后以30℃/min升至180℃、200℃、220℃、240℃或260℃,并分别保持恒温至分析结束。甘油乙醇溶液(WS-1、WS-2、WS-3)和葡萄酒(Wine-01)分别进样,每个试样进样3次,甘油经GC-C-IRMS转化的CO2的信号强度(m/z44)见图2,δ13C结果见表2。

图2 不同色谱柱最终温度时甘油转化的CO2的信号强度(m/z44)
由图2可知,各试样中甘油转化的CO2信号强度(m/z44)随色谱柱最终温度的升高而增大,最大相差近4倍,这表明柱温影响了甘油流出色谱柱后的分配情况,温度越高,越有利于甘油进入燃烧管。
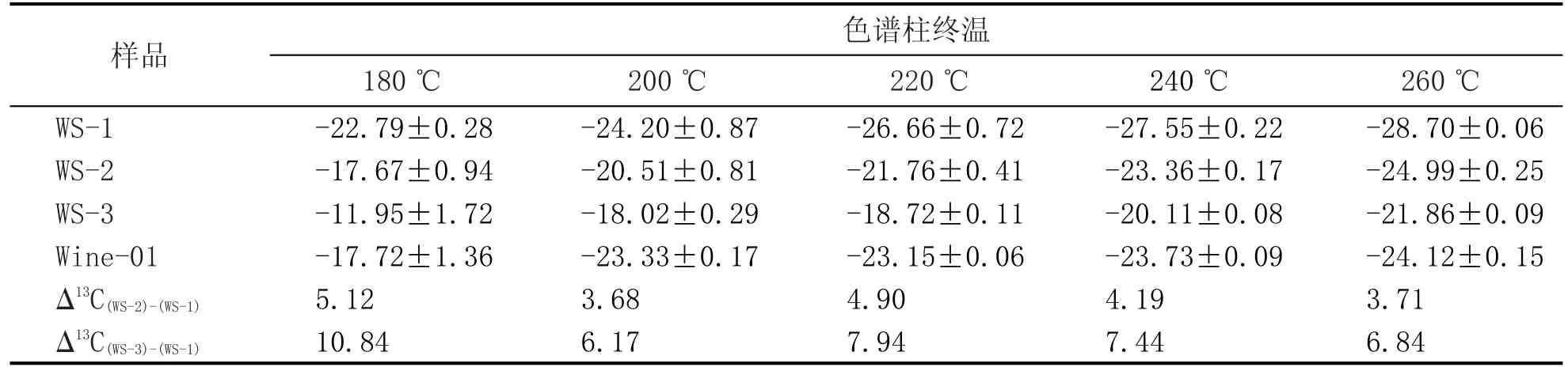
表2 甘油在不同色谱柱温度下的δ13C测定结果 (‰)
表2中,180~220℃时甘油δ13C测定值的标准偏差较大(>0.4‰),分析其原因,可能是由于甘油的沸点较高,甘油流出色谱柱后或在色谱柱中有一部分被液化的缘故。根据同位素热力学分馏原理,温度越高时同位素越能快速达到动态平衡,甘油在柱温180℃和200℃时,流入反应管的部分与剩余部分可能未达到平衡状态,因此导致碳同位素测定精度差。240℃和260℃时甘油δ13C的测定重复性均比较理想(标准偏差小于0.25‰),但相对于240℃,柱温设定在260℃时甘油标准品(乙醇为溶剂)产生的CO2的信号强度(m/z44)提升有限(2%~5%),而葡萄酒样品中甘油的信号强度(m/z44)提高了约9%。
稳定同位素分析领域,分析系统的测定尺度(“scale”)对分析准确性至关重要[23],而由于稳定同位素分析误差的存在(δ13C≤0.3‰),我们认为测定尺度越大,分析误差对样品准确性的影响越小(比如针对两个物质的测定尺度:实际差值=2,即使分析误差为0.3‰,那么数据经校正后结果的误差则可能仅为0.15‰)。标品WS-1与WS-3之间δ13CVPDB差值为5.90‰,180℃和220℃时差值最大,但分析精度差;与260℃的数据相比,240℃时GC-CIRMS的测定尺度更大一些,虽然260℃更有利于提升含水试样中甘油信号强度,但仍选择240℃作为后续研究的色谱柱的最终温度。
2.4 水分对甘油测定的影响
以上研究过程中,葡萄酒和甘油-乙醇溶液在测定时表现出了一些不同,其原因可能是由于样品中溶剂不同导致的:纯甘油样品的溶剂为纯乙醇,而葡萄酒中则是10%左右的乙醇水溶液。为研究水分对甘油测定的影响,分别配制含水量为0、20%、40%、60%、80%、90%和100%的乙醇水溶液,然后分别稀释甘油工作标准物质(WS-1和WS-3)至6 g/L,并在进样口270℃、流速1.2 mL/min、程序升温(80℃/1 min//30//240℃/5 min)条件下进样测定,每个试样测定3次,结果见图3和表3。

图3 不同溶剂中甘油转化CO2的信号强度(m/z44)
由图3可知,CO2信号强度均随溶液中水分含量的增加而降低,拟合度分析表明相关系数R2可达0.98,这表明溶液中水分影响了甘油在GC中的分配特征。事实上,同体积同条件进样,水蒸汽的体积是乙醇蒸汽的3倍以上,因此,进样溶液含水量越高,甘油气化后占比越低,进入色谱柱的气态甘油也会相应减少。
表3中所有试样的标准偏差均小于0.21‰,这说明水分未影响甘油δ13C的测定稳定性。但是各试样中甘油δ13C的分析结果存在微小差异,测定值随溶液中含水量的增加而升高,样品中的水分影响了甘油的碳同位素分馏情况,此时,进样口处含13C较多的甘油分子更容易被转移至色谱柱中。葡萄酒的含水量一般为84%~95%,对比表3数据可知,样品含水量在80%~100%波动时,两个甘油(WS-1和WS-2)δ13C测定值的极差分别为0.49‰和0.89‰,因此,按照稳定同位素分析的“IT”原则,应确保工作标准样品与葡萄酒样品尽可能具有相同的水/乙醇比例。2.5甘油浓度对δ13C测定的影响

表3 不同溶剂中甘油δ13C测定结果 (‰)

表4 不同浓度下甘油δ13C测定结果 (‰)
Hall等指出分析单体化合物的碳同位素分布特征时δ13C测定结果受浓度的影响较大[21],然而葡萄酒甘油含量因受多种因素的影响而存在差异,有报道指出葡萄酒中甘油含量最低时仅为4 g/L,而最高则可达21 g/L[2]。为研究甘油浓度对δ13C测定的影响,我们模拟葡萄酒中酒精含量,以12%乙醇水溶液为溶剂配制浓度为3~21 g/L的甘油样品,分别测定δ13C,并以100%乙醇为溶剂配制的甘油样品为基准进行数据校正,结果见表4。
由表4可看出,峰面积与甘油浓度呈显著正相关关系(R2=0.978),然而当甘油浓度加倍时,甘油转化CO2的m/z 44峰面积的增加量却不止一倍。对比分析精度,除浓度为3 g/L时测定结果稳定性SD大于0.4‰以外,其余浓度梯度时SD均优于0.3‰,但是甘油浓度在12 g/L时的样品与相邻两个浓度样品的测定结果具有明显差异,这意味着测定结果在甘油浓度9~15 g/L时存在较大变化,而甘油浓度在6~9 g/L与15~21 g/L范围时,δ13C测定值则保持了相对稳定(差异均小于0.2‰)。一般来说,连续流-稳定同位素比值质谱仪(CF-IRMS)测定CO2时,δ13C测定结果会因信号强度(m/z44)的不同而线性变化,根据仪器公司提供的测试指标要求,变化率在±0.065‰/V范围内时即可认为仪器测定结果是稳定的。本研究中,3 g/L(信号强度为502 mV)时,δ13C测定结果相对偏正,高浓度21 g/L(信号强度为13994 mV)时,结果最偏负,虽然变化是线性的,但是3个甘油工作标准的变化率不一致,分别为-0.084‰/V~-0.117‰/V,其中甘油中13C含量越高变化率越大,说明GC-C-IRMS分析甘油δ13C时具有特殊的碳同位素分馏效应,而且浓度确实造成了δ13C测定结果的差异。
当然,根据“IT”原则可在测定结果稳定的前提下进行数据校正,然后依据“IT”原则,用于校正的标准品应与待测样品具有相同的理化特征和处理过程。采用GC-C-IRMS分析葡萄酒甘油时应确保标准品的甘油浓度和乙醇含量完全一致(或接近),然而葡萄酒乙醇含量为8%vol~20%vol,而甘油浓度在3~21 g/L之间波动,并且不同产区、不同品种的葡萄酒样品上述两指标均存在较大差异,为确保数据有效性应根据单个样品进行针对性的配制标准品,而这间接地影响了分析效率,增加了前处理和分析工作量。基于此,不建议使用GCC-IRMS技术测定葡萄酒中甘油碳同位素比值。
3 结论
本研究分析了温度、样品含水量及甘油浓度对GC-C-IRMS测定甘油δ13C的影响,其中甘油产生的CO2的信号强度(m/z44)与温度、浓度呈显著正相关关系,然而与样品含水量呈显著负相关;甘油δ13C测定值与温度呈显著负相关而与样品含水量呈显著正相关,但与浓度的相关性不明显;各条件下3个工作标准的测定结果与标定值(EA数据)具有良好相关性,可根据数据模型用于数据校正,但前提是用于校正的标准品与待测样品的乙醇和甘油浓度一致,而该过程会直接增加前处理工作量和测定任务量,分析效率低下,因此不建议使用GC-CIRMS技术测定葡萄酒中甘油碳同位素比值。
参考文献:
[1]NIEUWOUDT H,PRIOR B,PRETORIUS I,et al. Glycerol in South African table wines:an assessment of its contribution to wine quality[J].S Afr j enol vitic,2002,23:22-30.
[2]CALDERONE G,NAULET N,GUILLOU C,et al.Characterization of European wine glycerol:stable carbon isotope approach[J].Journal of agricultural and food chemistry,2004,52(19):5902-5906.
[3]OGRINC N,KOŠIR I J,SPANGENBERG J E,et al.The application of NMR and MS methods for detection of adulteration of wine,fruit juices,and olive oil:a review[J].Analytical and bioanalytical chemistry,2003,376(4):424-430.
[4]WEBER D,KEXEL H,SCHMIDT H L.13C-pattern of natural glycerol:origin and practical importance[J].Journal of agricultural and food chemistry,1997,45(6):2042-2046.
[5]ROSSMANN A,SCHMIDT H L,HERMANN A,et al.Multielement stable isotope ratio analysis of glycerol to determine its origin in wine[J].Zeitschrift für Lebensmitteluntersuchung und-Forschung A,1998,207(3):237-243.
[6]FRONZA G,FUGANTI C,GRASSELLI P,et al.Determination of the13C content of glycerol samples of different origin[J].Journal of agricultural and food chemistry,1998,46(2):477-480.
[7]FRONZA G,FUGANTI C,GRASSELLI P,et al.δ13C-and δ18O-Values of glycerol of food fats[J].Rapid communications in mass spectrometry,2001,15(10):763-766.
[8]陈珊珊,钟其顶,俞慧红,等.植物油中甘油稳定碳同位素组成的测定[J].质谱学报,2016,37(4):359-365.
[9]陈文斌,钟其顶,王道兵,等.气相色谱-同位素质谱(GC-C-IRMS)测定饮料酒中乙醇13C/12C比值方法研究[J].酿酒科技,2013(5):27-30.
[10]钟其顶,王道兵,孟镇,等.有机溶剂稀释与气相色谱-燃烧-同位素质谱(GC-C-IRMS)联用测定食醋中乙酸的δ13C[J].质谱学报,2014,35(4):372-377.
[11]CALDERONE G,NAULET N,GUILLOU C,et al.Characterization of European wine glycerol:stable carbon isotope approach[J].Journal of agricultural and food chemistry,2004,52(19):5902-5906.
[12]WANG D,ZHONG Q,LI G,et al.Rapid method for the determination of the stable oxygen isotope ratio of water in alcoholic beverages[J].Journal of agricultural and food chemistry,2015,63(42):9357-9362.
[13]ELSNER M,JOCHMANN M A,HOFSTETTER T B,et al.Current challenges in compound-specific stable isotope analysis of environmental organic contaminants[J].Analytical and bioanalytical chemistry,2012,403(9):2471-2491.
[14]WERNER R A,BRAND WA.Referencing strategies and techniques in stable isotope ratio analysis[J].Rapid communications in mass spectrometry,2001,15(7):501-519.
[15]CARTER J F,FRY B.Ensuring the reliability of stable isotope ratio data–beyond the principle of identical treatment[J].Analytical and bioanalytical chemistry,2013,405(9):2799-2814.
[16]KRUMMEN M,HILKERTA W,JUCHELKA D,et al.A new concept for isotope ratio monitoring liquid chromatography/mass spectrometry[J].Rapid communications in mass spectrometry,2004,18(19):2260-2266.
[17]HATTORI R,YAMADA K,SHIBATA H,et al.Measurement of the isotope ratio of acetic acid in vinegar by HS-SPME-GC-TC/C-IRMS[J].Journal of agricultural and food chemistry,2010,58(12):7115-7118.
[18]王旭,张福松,丁仲礼.EA-Conflo-IRMS联机系统的燃烧转化率漂移及其对氮,碳同位素比值测定的影响[J].质谱学报,2006,27(2):104-109.
[19]SCHMITT J,GLASER B,ZECH W.Amountdependent isotopic fractionation during compoundspecific isotope analysis[J].Rapid communications in mass spectrometry,2003,17(9):970-977.
[20]ELLEHOJ M D,STEEN-LARSEN H C,JOHNSEN S J,et al.Ice-vapor equilibrium fractionation factor of hydrogen and oxygen isotopes:experimental investigations and implications for stable water isotope studies[J].Rapid communications in mass spectrometry,2013,27(19):2149-2158.
[21]HALL J A,BARTH J A C,KALIN R M.Routine analysis by high precision gas chromatography/mass selective detector/isotope ratio mass spectrometry to 0.1 parts per mil[J].Rapid communications in mass spectrometry,1999,13(13):1231-1236.
[22]刘青,刘朝霞,李志勇,等.国产葡萄酒中甘油含量的调查与分析[J].中国食品卫生杂志,2015,27(2):171-175.
