苯磺酰胺类IDO1抑制剂的设计、合成和生物活性
2018-03-20金双龙郭文洁赖宜生
金双龙,王 芳,邹 毅,王 燕,胡 月,郭文洁,徐 强*,赖宜生**
(1中国药科大学新药研究中心,江苏省代谢性疾病药物重点实验室,南京 210009;2南京大学生命科学学院,医药生物技术国家重点实验室,南京 210093)
吲哚胺2,3-双加氧酶1(IDO1)是人体肝脏外催化色氨酸沿犬尿氨酸途径代谢的起始和限速酶。研究表明,IDO1通过催化色氨酸代谢从而对机体的固有免疫和适应性免疫起重要的调节作用[1]。在肿瘤微环境中,肿瘤细胞可以通过诱导IDO1的过度表达,造成局部色氨酸耗竭和犬尿氨酸等代谢物的聚积,从而通过激活GCN2和AHR信号通路抑制T细胞的增殖及诱导其凋亡,并且刺激初始T细胞分化成调节性T细胞,进而介导肿瘤免疫逃逸[2]。因此,IDO1已成为目前肿瘤免疫治疗小分子药物研发的热门靶点。
近年来,科研人员通过天然产物、高通量筛选和虚拟筛选等途径陆续发现了醌类、吲哚类、咪唑类、呋咱羟脒类等结构类型的IDO1抑制剂。其中,Ueng课题组[3]于2014年报道了一系列苯磺酰肼类IDO1抑制剂,化合物U-3i对IDO1的抑制活性最强,在基于HeLa细胞的酶活测试中其EC50为172 nmol/L。分子对接结果表明,除了疏水和氢键相互作用以外,化合物U-3i结构中磺酰基的一个氧原子还与血红素铁离子形成络合作用。
为了寻找新型IDO1抑制剂,本研究在化合物U-3i的结构基础上,考虑到肼类化合物潜在的毒性[4],通过生物电子等排原理,结合计算机模拟分子对接技术,设计并合成了一系列苯磺酰胺类目标化合物(图1),在考察中间连接链的基础上,侧重探索了伸向IDO1活性口袋A和B的两个苯环上取代基对活性的影响。体外活性测试结果表明,化合物3b和3e对基于HeLa细胞的IDO1活性显示出了较好的抑制作用。进一步实验表明,化合物3b和3e能够促进T细胞的增殖,诱导γ干扰素(IFN-γ)的生成,并且能够抑制调节性T细胞的分化,因此具有进一步研究的价值。

Figure1 Design of target compounds
1 合成路线
各种取代的苯磺酰氯与取代苯胺或苄胺进行缩合反应生成相应的苯磺酰胺3a~3h。其中,4-乙酰胺基苯磺酰胺衍生物(3b~3d)在浓盐酸的存在下经脱乙酰化得到4-胺基苯磺酰胺盐酸盐,最后经碳酸钾中和即制得4-胺基苯磺酰胺衍生物4a~4c(路线1)。所有目标化合物的结构均经MS和1H NMR确证。
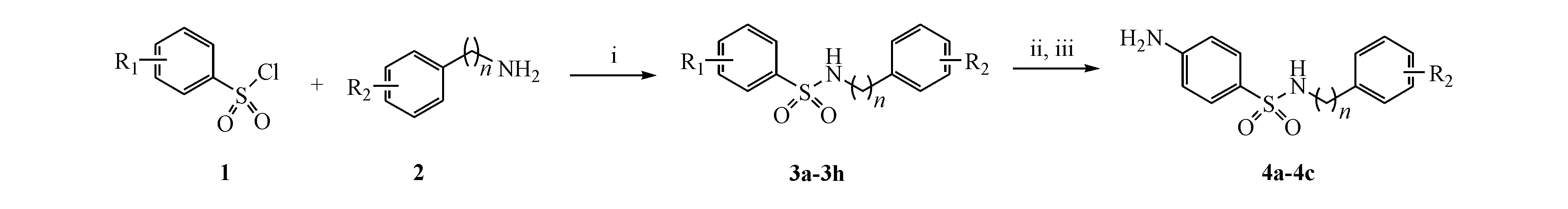
Scheme1 Reagents and conditions:(i) TEA,DCM,r.t.,5 h;(ii) HCl,ethanol,reflux,12 h;(iii) K2CO3
2 实验部分
2.1 仪器和试剂
熔点采用RY-1型熔点仪测定,温度计未经校正;1H NMR使用ACF-300 MHz型核磁共振仪测定,TMS为内标;MS使用Agilent 1100 Series LC/MSD Trap(SL)质谱仪测定。所有试剂未经特别说明均为市售化学纯或分析纯产品。
2.2 化学合成
N-(4-(N-(4-氰苄基)氨磺酰)苯基)乙酰胺(3a)的合成 将4-氰基苯乙胺(1.00 g,7.57 mmol)溶于无水二氯甲烷(15 mL)中,加入三乙胺(1.26 mL,8.75 mmol),冰浴下滴入含4-乙酰氨基苯磺酰氯(1.93 g,8.32 mmol)的无水二氯甲烷溶液5 mL,滴毕于冰浴中搅拌30 min,撤去冰浴,室温反应4 h,加水25 mL,二氯甲烷萃取,无水硫酸镁干燥,柱色谱(EA-PE,1∶5)纯化得到白色固体2.18 g,收率87%,mp 212~213 ℃。1H NMR(300 MHz,DMSO-d6)δ:10.31(1H,s,CONH),8.18(1H,s,NH),7.71(6H,q,J=9.8,8.8 Hz,Ar-H),7.44(2H,dd,J=8.3,4.3 Hz,Ar-H),4.05(2H,d,J=7.0 Hz,CH2),2.25~1.99(3H,m,CH3);ESI-MS:m/z328.0[M-H]-。
用类似的方法制备得到目标化合物3b~3h。
N-(4-(N-(4-甲氧基苄基)氨磺酰)苯基)乙酰胺(3b) 白色固体,收率80%,mp 188~190 ℃。1H NMR(300 MHz,DMSO-d6)δ:10.38(1H,s,CONH),7.96(1H,s,NH),7.76~7.68(4H,m,Ar-H),7.13(2H,d,J=8.3 Hz,Ar-H),6.83(2H,d,J=8.3 Hz,Ar-H),3.86(2H,s,CH2),3.71(3H,s,OCH3),2.09(3H,s,CH3);ESI-MS:m/z333.0[M-H]-。
N-(4-(N-(4-甲氧基苯基)氨磺酰)苯基)乙酰胺(3c) 白色固体,收率88%,mp 206~208 ℃。1H NMR(300 MHz,DMSO-d6)δ:10.29(1H,s,CONH),9.77(1H,s,NH),7.78~7.49(4H,m,Ar-H),6.95(2H,dq,J=8.2,3.2 Hz,Ar-H),6.86~6.70(2H,m,Ar-H),3.66(3H,q,J=2.2 Hz,OCH3),2.06(3H,q,J=3.5 Hz,CH3);ESI-MS:m/z319.0[M-H]-。
N-(4-(N-(4-氟苯基)氨磺酰)苯基)乙酰胺(3d) 白色固体,收率91%,mp 190~192 ℃。1H NMR(300 MHz,DMSO-d6)δ:10.32(1H,d,J=5.5 Hz,CONH),10.13(1H,s,NH),7.78~7.56(4H,m,Ar-H),7.20~6.97(4H,m,Ar-H),2.06(3H,d,J=5.2 Hz,CH3);ESI-MS:m/z307.0[M-H]-。
N-(4-氰苄基)-3-硝基苯磺酰胺(3e) 红色固体,收率90%,mp 153~155 ℃。1H NMR(300 MHz,DMSO-d6)δ:8.85~8.32(3H,m,Ar-H),8.14(1H,s,NH),7.99~7.61(3H,m,Ar-H),7.39(2H,s,Ar-H),4.16(2H,s,CH2);ESI-MS:m/z316.0[M-H]-。
N-(4-甲氧基苄基)-3-硝基苯磺酰胺(3f) 红色固体,收率80%,mp 103~105 ℃。1H NMR(300 MHz,DMSO-d6)δ:8.45(1H,s,NH),8.37(1H,ddd,J=8.3,2.3,1.0 Hz,Ar-H),8.30(1H,t,J=2.0 Hz,Ar-H),8.10(1H,ddd,J=7.9,1.8,1.0 Hz,Ar-H),7.79(1H,t,J=8.0 Hz,Ar-H),7.09~7.00(2H,m,Ar-H),6.76~6.65(2H,m,Ar-H),3.98(2H,s,CH2),3.64(3H,s,CH3);ESI-MS:m/z321.0[M-H]-。
N-(4-甲氧基苯基)-3-硝基苯磺酰胺(3g) 红色固体,收率86%,mp 130~132 ℃。1H NMR(300 MHz,CHCl3-d)δ:8.61(1H,t,J=2.0 Hz,Ar-H),8.43(1H,ddd,J=8.3,2.2,1.1 Hz,Ar-H),7.99(1H,ddd,J=7.8,1.8,1.1 Hz,Ar-H),7.68(1H,t,J=8.0 Hz,Ar-H),7.06~6.98(2H,m,Ar-H),6.87~6.71(2H,m,Ar-H),6.52(1H,s,NH),3.80(3H,s,CH3);ESI-MS:m/z307.0[M-H]-。
4-氰基-N-(4-甲氧基苄基)苯磺酰胺(3h) 白色固体,收率75%,mp 137~139 ℃。1H NMR(300 MHz,CHCl3-d)δ:7.99~7.90(2H,m,Ar-H),7.85~7.75(2H,m,Ar-H),7.15~7.04(2H,m,Ar-H),6.86~6.75(2H,m,Ar-H),4.94(1H,t,J=5.9 Hz,NH),4.16(2H,d,J=5.9 Hz,CH2),3.80(3H,s,CH3);ESI-MS:m/z301.0[M-H]-。
4-氨基-N-(4-甲氧基苯基)苯磺酰胺(4a) 将3c(0.50 g,1.56 mmol)溶于乙醇(10 mL)中,加入浓盐酸(3 mL),回流12 h,冷却至室温,加入饱和碳酸钾溶液将反应液pH调到9。减压除去乙醇,乙酸乙酯萃取,无水硫酸镁干燥,减压蒸除溶剂,得到白色固体0.42 g,收率97%,mp 131~133 ℃。1H NMR(300 MHz,DMSO-d6)δ:9.48(1H,s,NH),7.40~7.19(2H,m,Ar-H),7.06~6.86(2H,m,Ar-H),6.84~6.70(2H,m,Ar-H),6.59~6.42(2H,m,Ar-H),5.95(2H,s,NH2),3.66(3H,d,J=2.3 Hz,CH3);ESI-MS:m/z277.0[M-H]-。
用类似的方法制备得到目标化合物4b~4c。
4-氨基-N-(4-氟苯基)苯磺酰胺(4b) 白色固体,收率78%,mp 151~153 ℃。1H NMR(300 MHz,DMSO-d6)δ:9.81(1H,s,NH),7.38~7.29(2H,m,Ar-H),7.06(4H,dd,J=7.0,3.2 Hz,Ar-H),6.52(2H,dd,J=8.9,3.1 Hz,Ar-H),5.99(2H,s,NH2);ESI-MS:m/z265.0[M-H]-。
4-氨基-N-(4-甲氧基苄基)苯磺酰胺(4c) 白色固体,收率75%,mp 94~96 ℃。1H NMR(300 MHz,DMSO-d6)δ:7.54(1H,s,NH),7.43(2H,d,J=8.3 Hz,Ar-H),7.19~7.09(2H,m,Ar-H),6.91-6.79(2H,m,Ar-H),6.65~6.54(2H,m,Ar-H),5.94(2H,s,NH2),3.79(2H,s,CH2),3.72(3H,s,CH3);ESI-MS:m/z291.0[M-H]-。
2.3 生物活性评价
2.3.1 基于HeLa细胞的酶活性评价 按每孔5×103个细胞的密度将HeLa细胞接种在96孔培养板中,贴壁后加入IFN-γ(100 ng/mL)处理24 h,吸去上清液,加入L-色氨酸(15 μg/mL)、IFN-γ(100 ng/mL)、受试物至200 μL体系,设置空白对照、模型组和药物处理组(以GDC-0919[5]为阳性对照),培养24 h,收集上清液140 μL,加入6.1 mol/L三氯乙酸10 μL,50 ℃温育30 min,离心去除沉淀物。将各孔上清液100 μL移至另一96孔板中,与等体积20 mg/mL对-二甲氨基苯甲醛的乙酸溶液混合。使用酶标仪在480 nm处检测吸收度,计算化合物的IC50。
2.3.2 T细胞转化实验 将处理过的B16F1细胞(每孔8×104个细胞)、小鼠脾脏淋巴细胞(每孔1×106个细胞,使用5 μg/mL ConA刺激)加入24孔板中,加入对应浓度的化合物后置于37 ℃、湿度95%、5% CO2的培养箱中孵育48 h。一方面,加入MTT(终浓度4 mg/mL)20 μL培养箱中继续孵育4 h,酶标仪测定570 nm波长处吸收度,计算T细胞增殖率;另一方面,收集上清液使用ELISA检测细胞因子IFN-γ含量,同时使用抗CD4、抗CD25、抗FOCP3抗体染色,在流式细胞仪中检测T细胞的分化。
2.4 分子模拟
利用Schrödinger软件包(2009)中的Protein and Ligand Preparation模块在基于OPLS-2005的力场条件下对蛋白加氢、加电荷和质子化处理,并进行能量优化;小分子使用Ligprep模块进行配体准备。利用Gold进行分子对接,采用含血红素参数的GoldScore打分函数,此参数是由PDB数据库经统计分析得到[6]。以血红素亚铁离子为中心,其10 Å半径内氨基酸残基作为对接的活性位点。在对接中使用遗传算法,运行10次。范德华和氢键参数的截断值分别设为4.0和2.5 Å。保留打分最高的结合模式作为最终对接结果用于分析。
3 结果与讨论
体外基于HeLa细胞的酶活性检测结果如表1所示,大部分化合物对IDO1都显示出一定的抑制活性,其中化合物3b和3e的活性较强,其IC50分别为5.88和10.25 μmol/L。
Table1 Inhibitory activity of the target compounds against IDO1

CompdR1R2nIC50/(μmol/L)3ap⁃CH3CONHp⁃CN154083bp⁃CH3CONHp⁃OCH315883cp⁃CH3CONHp⁃OCH3057283dp⁃CH3CONHp⁃F0>1003em⁃NO2p⁃CN110253fm⁃NO2p⁃OCH3133753gm⁃NO2p⁃OCH3042613hp⁃CNp⁃OCH3137934ap⁃NH2p⁃OCH3038624bp⁃NH2p⁃F0>1004cp⁃NH2p⁃OCH31>100GDC⁃0919035
为了了解化合物3b和3e对T细胞增殖和调节性T细胞分化的影响情况,本研究利用高表达IDO1的B16F1细胞与小鼠脾脏淋巴细胞共培养体系开展了T细胞转化实验,结果如图2所示。一方面,在混合细胞体系中,B16F1细胞会对T细胞的增殖产生抑制作用,而化合物3b和3e能在一定程度上增加T细胞的增殖,其中,化合物3e能使T细胞的增殖率提高35%(图2-A),而且化合物3b和3e能够促进IFN-γ的生成(图2-B)。另一方面,从图2-C和图2-D可以看出,当初始T细胞与B16F1细胞共培养时,调节性T细胞的数量与对照组相比增加了4倍多,而化合物3b和3e与阳性药GDC-0919一样都能明显抑制调节性T细胞的分化。这些实验结果提示,化合物3b和3e能够通过抑制IDO1来逆转其介导的免疫抑制。
初步构效关系分析表明,从中间连接链来看,与苯胺衍生物(3c,3d,3f)相比,苄胺衍生物(3a,3b,3g)展现出更强的活性,尤其是化合物3b(IC50:5.88 μmol/L)的活性比化合物3c(IC50:57.28 μmol/L)提高近10倍,说明苄胺相对于苯胺更有利于活性的提高(4a和4c除外)。从伸向B口袋的苯环上的取代基来看,含有供电子基的化合物活性普遍强于相应的吸电子基(3b优于3a,3c优于3d,4a优于4b,但3f不如3e)。有趣的是,从A口袋的苯环上的取代基来看,如果将4-乙酰胺基水解成胺基时活性明显下降(4a除外),而且从含有硝基和氰基的3e~3h来看,它们都具有较好的活性,提示A口袋苯环上的吸电子基比强供电子基(胺基)可能更有利于活性的提高。

*P< 0.05vsT+B16F1 group
为了进一步说明化合物的构效关系,本研究使用分子对接方法研究了化合物3b在IDO1活性位点中的结合模式。从图3可以看出,苯磺酰胺片段能很好地深入口袋A内部,并与Phe163产生π-π相互作用,而苄胺片段则伸向由Phe163,Phe226,Arg231,Leu234和Ile354形成的B口袋。此外,磺酰基中的一个氧原子与血红素亚铁产生络合作用,而另一个氧原子则与loop区Ala264 NH形成氢键作用。这些信息为本研究后续开展结构优化提供了理论指导。

Figure3 Proposed binding mode of compound3bdocked into the crystal structure of human IDO1(PDB ID:4PK5)
[1] Uyttenhove C,Pilotte L,Théate I,etal.Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase[J].NatMed,2003,9(10):1269-1274.
[2] Platten M,von Knebel Doeberitz N,Oezen I,etal.Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors[J].FrontImmunol,2015,5:673.
[3] Cheng MF,Hung MS,Song JS,etal.Discovery and structure-activity relationships of phenyl benzenesulfonylhydrazides as novel indoleamine 2,3-dioxygenase inhibitors[J].BioorgMedChemLett,2014,24(15):3403-3406.
[4] Nelson SD.Metabolic activation and drug toxicity[J].JMedChem,1982,25(7):753-765.
[5] Zou Y,Wang F,WANG Y,etal.Discovery of imidazoleisoindole derivatives as potent IDO1 inhibitors:design,synthesis,biological evaluation and computational studies[J].EurJMedChem,2017,140:293-304.
[6] Jones G,Willett P,Glen RC,etal.Development and validation of a genetic algorithm for flexible docking[J].JMolBiol,1997,267(3):727-748.
