c-kit突变型与野生型胃肠道间质瘤中基因表达谱的鉴定
2018-03-01王春萌杨凌舸师英强
陈 杰,王春萌,罗 鹏,杨凌舸,师英强
1. 复旦大学附属肿瘤医院胃外科,复旦大学上海医学院肿瘤学系,上海 200032;
2. 复旦大学附属肿瘤医院骨与软组织外科,复旦大学上海医学院肿瘤学系,上海 200032
胃肠道间质瘤(gastrointestinal stromal tumor,GIST)是消化道最常见的间叶来源肿瘤,主要发生于胃和小肠,少数发生于腹膜后、网膜和肠系膜,约占全部胃肠道恶性肿瘤的2%[1]。近年来,GIST的概念提出后,其诊断和治疗有了飞速发展,这得益于GIST中特征性遗传改变被发现c-kit基因突变[1]。目前伊马替尼是转移性或不可切除GIST标准的一线治疗药物[7]。但是,越来越多的研究表明,伊马替尼在治疗GIST过程中存在原发性耐药和继发性耐药,且耐药机制复杂,其中原因可能与肿瘤中存在继发c-kit基因突变的多细胞克隆有关。因此,对于伊马替尼原发或继发耐药的晚期GIST患者的治疗目前仍是一个难题。在这一背景下,寻找新的致病因子及治疗靶点,从而为胃肠道间质瘤的治疗提供新的策略和思路显得十分重要。
c-kit基因位于人染色体4q12-13上,编码Ⅲ型跨膜生长因子受体。其受体蛋白由细胞内酪氨酸蛋白结构域、跨膜结构域及细胞外配体结构域共同组成。已有研究指出,c-kit在多种肿瘤中表达异常,其在多种实体肿瘤中表达上调,但在乳腺癌、黑色素瘤及甲状腺癌中呈低表达[2]。在肝癌中,TGF-β/c-kit能够形成一个正反馈环促进肿瘤的发生、发展[3]。但是,对于c-kit在肿瘤中的作用,我们仍然知之甚少。国内外研究表明,c-kit基因突变率为75%~80%[3]。其中最常见的是外显子11突变(65%~70%),以及外显子9突变(10.0%)、外显子13(1.7%)和外显子17(1.3%)等少见突变位点。突变类型以缺失突变、点突变、混合突变和插入突变为主[4]。靶向药物甲磺酸伊马替尼治疗GIST的机制在于, 当c-kit激酶激活后,使酪氨酸残基磷酸化,从而调节细胞的生长和凋亡[5]。
除了由基因突变引起的GIST外,还有一类特殊GIST,即野生型GIST。实际这个命名并非完全准确,而是对形态学符合GIST,CD117阳性或阴性表达,同时未能检测到c-kit和血小板源性生长因子受体α(platelet-derived growth factor receptor alpha,PDGFRA)基因突变的GIST的统称。国内外研究发现,85%的儿童GIST和10%~15%的成人GIST既没有c-kit基因突变,也没有PDGFRA基因突变,故被称为野生型[6]。到目前为止,野生型GIST的发病机制仍不明确,它并非是单一的GIST种类,而是一个由生物特性迥异的多个亚群组成的家族,其基因分子发病机制也可能不完全相同,这一领域亟需进一步深入研究。
在本研究中,我们采用4对原发性GIST与相应的癌旁组织,去除核糖体RNA后,构建RNA测序文库,采用美国Illumina公司Hiseq 2500进行双端测序。结合生物信息分析,用TopHat2得到c-kit突变型与野生型GIST中差异表达的基因。希望通过探索c-kit突变对于GIST组织表达谱的影响,从而为研究GIST的发病机制提供新的理论依据,同时也尝试为GIST的治疗提供新的策略和思路。
1 材料和方法
1.1 组织样本
4对GIST及其癌旁组织均来自复旦大学附属肿瘤医院胃外科和骨与软组织外科手术切除标本,获取标本均得到伦理委员会的同意,并经过了严格病理诊断。
1.2 组织总RNA的抽提
采用TRIzol试剂(购自美国Invitrogen公司)进行组织总RNA的抽提。每毫升TRIzol中加入200 μL的氯仿萃取,将无色上清液转移带一个新的离心管中,加入等体积的异丙醇沉淀并以8 000×g的速率离心10 min,并用75%乙醇洗涤。弃上清液,室温干燥RNA沉淀,用DEPC处理过H2O溶解沉淀,定量备用。
1.3 RNA测序
取3 μg总RNA,使用RiboMinus Eukaryote Kit(购自德国Qiagen公司)去除核糖体RNA,随后用NEBNext Ultra Directional RNA Library Prep Kit for Illumina(购自美国NEB公司)构建RNA测序文库。文库质检后,在Illumina NEX seq 500机器上进行测序。
1.4 统计学处理
差异表达基因热图、火山图、GO富集分析及KEGG信号通路分析均使用R(3.3.2)软件进行分析,分别使用gplots、ggplot2和clusterProfiler pachage完成。
2 结 果
2.1 RNA测序预测受c-kit调控的基因
我们利用RNA高通量测序结合生物信息学技术对c-kit突变型与野生型的胃肠道间质瘤组织进行大规模测序。首先,从4例c-kit野生型患者和4例c-kit突变型患者组织中抽提总RNA,并去除了其中的核糖体RNA。每个样本都在Illumina HiSeq 2000测序仪上进行RNA测序,并产生了约60兆的读段。利用TopHat2软件,我们将这些读段映射到人类参考基因组中(GRCh37/hg19)。
此外,本研究分析了其中的mRNA,分析了c-kit突变型与野生型患者中表达谱的差异,发现其中有263个基因差异有统计学意义(图1)。c-kit突变型与野生型中表达谱的差异提示,这263个基因可能受到c-kit基因的调控。
2.2 基因富集分析
为了探讨c-kit突变对于GIST的影响,本研究对上述263个基因进行了GO和KEGG通路富集分析。GO分析中将差异表达的基因按功能分为3大类:生物学过程(biological process,BP)、分子功能(molecular function,MF)和细胞成分(cellular component,CC)。GO分析结果显示,这263个基因显著富集于组蛋白分解过程、蛋白激酶激活剂及T细胞受体信号小体中(图2,表1)。c-kit突变型与野生型GIST组织中,差异基因在KEGG数据库中富集于Hedgehog信号通路、FoxO信号通路、过氧化物酶体、胆汁酸合成以及谷氨酰胺和谷氨酸的代谢通路中(图2,表2)。
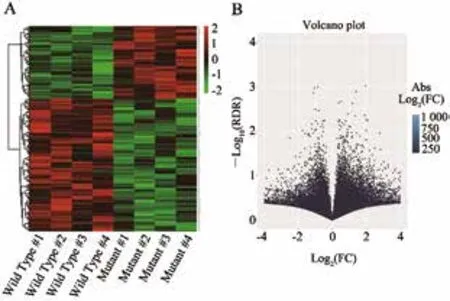
图 1 c-kit 突变型与野生型的胃癌组织中差异表达基因Fig. 1 Differentially expressed genes of c-kit mutant and wild type GIST tissues

图 2 GO和KEGG通路分析Fig. 2 GO and KEGG pathway analysis

表 1 表达差异的基因功能Tab. 1 The functions of the expression-differentiated genes

表 2 KEGG通路数据库富集分析Tab. 2 The enrichment analysis of KEGG pathway database
3 讨 论
GIST能够发生在消化系统中的许多位置,是最常见的胃肠道间叶源性肿瘤,常发生于肠系膜、胃等组织中,少量发生于十二指肠和直肠中。受体酪氨酸激酶c-kit和PDGFRA的突变被认为是GIST的主要发病因素和推动因素[1,4]。在GIST中c-kit具有高频突变,其中常见的是第11号外显子、第9号外显子和第17号外显子[5]。c-kit编码的蛋白产物CD117的发现提供了一个更加敏感和特异的诊断指标[6]。基因突变类型能够决定靶向药物治疗的敏感性。因此,一般推荐对患者术后的GIST标本进行常规基因检测。c-kit和PDGFRA的突变信息能够显著提升我们对于患者预后信息的判断。在c-kit发生高频突变患者中更容易发生复发转移,也具有更差的无瘤生存[7]。同时,c-kit能够作用GIST的治疗靶点。随着GIST靶向治疗的广泛开展,c-kit基因突变与靶向治疗的关系已基本达成共识。研究最为集中的是GIST患者基因突变类型与目前最常用的一线靶向治疗药物伊马替尼。伊马替尼可通过竞争性结合受体酪氨酸激酶的ATP结合位点,抑制c-kit和PDGFRA的自身磷酸化,进而抑制c-kit和PDGFRA下游信号分子的异常激活。所以,通过了解c-kit在GIST分子层面上的影响,对进一步阐明GIST发生和GIST靶向治疗机制具有至关重要的作用[8]。
本研究对4例有c-kit突变和4例没有c-kit突变的GIST组织进行测序,寻找其背后的差异基因。发现有263个基因在这两组患者肿瘤组织中有显著性差异。其中,170个基因在野生型患者组织中的表达量高于c-kit突变型患者组织;而93个基因由于c-kit突变而得到上调。利用这些基因分析c-kit突变引起的细胞信号通路与功能变化,进行GO分析,发现这些差异表达的基因广泛参与组蛋白mRNA代谢、组蛋白去甲基化、内质网应激及JNK信号通路等。KEGG信号通路分析发现,c-kit引起的差异表达基因富集在Hedgehog、FoxO信号通路及谷氨酰胺、谷氨酸代谢通路中。许多肿瘤的发生、发展伴随着如Hedgehog或FoxO信号通路的变化;反之,这些信号通路的变化也能调控肿瘤的发生、发展[9-10]。本研究提示,c-kit在GIST发生中具有一定的作用,但仍有许多问题亟待解决。在GIST中,c-kit的具体作用机制仍然需要更多的分子生物学实验与动物模型进行详细验证。
[1] HIROTA S, ISOZAKI K, MORIYAMA Y, et al. Gain-offunction mutations of c-kit in human gastrointestinal stromal tumors[J]. Science, 1998, 279(5350): 577-580.
[2] LESSI F, FRANCESCHI S, TANTILLO E, et al. Loss of c-kit in thyroid cancer cells: a functional study to investigate its role in tumor differentiation and progression[J]. Euro J Cancer,2016, 61: 82-83.
[3] ROJAS A, ZHANG P, WANG Y, et al. A positive TGF-β/c-kit feedback loop drives tumor progression in advanced primary liver cancer[J]. Neoplasia, 2016, 18(6): 371-386.
[4] HEINRICH M C, CORLESS C L, DUENSING A, et al.PDGFRA activating mutations in gastrointestinal stromal tumors[J]. Science, 2003, 299(5607): 708-710.
[5] LASOTA J, XI L, COATES T, DENNIS R, et al. No KRAS mutations found in gastrointestinal stromal tumors (GISTs):molecular genetic study of 514 cases[J]. Mod Pathol, 2013,26(11): 488-1491.
[6] BEHAM A W, SCHAEFER I M, SCHÜLER P, et al.Gastrointestinal stromal tumors[J]. Int J Colorectal Dis,2012, 27(6): 689-700.
[7] LASOTA J, DANSONKA-MIESZKOWSKA A, SOBIN L H, et al. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential[J].Lab Invest, 2004, 84(7): 874-883.
[8] 侯亚超, 邓靖宇, 梁 寒. 基因检测在胃肠间质瘤诊疗中的应用[J]. 中华胃肠外科杂志, 2015, 18(4): 305-308.
[9] JIANG J, HUI C. Hedgehog signaling in development and cancer[J]. Dev Cell, 2008, 15(6): 801-812.
[10] ARDEN K C. Multiple roles of FOXO transcription factors in mammalian cells point to multiple roles in cancer[J]. Exp Gerontol, 2006, 41(8): 709-717.
