微波法制备纳米碳点反应机制与发光机理
2018-01-29刘金龙林亮珍胡锦凤白明洁陈良贤魏俊俊黑立富李成明
刘金龙,林亮珍,胡锦凤,白明洁,陈良贤,魏俊俊,黑立富,李成明
1 引言
纳米碳点是近年来被广泛研究的一种新型荧光碳纳米粒子。通常被认为是一种近似球型且直径在10 nm以内的零维半导体纳米晶体,也被认为是一种由少量原子或分子组成的纳米团簇1-3。纳米碳点具有良好的光学特性,特别是在紫外光谱吸收较强,其吸收峰通常在260-320 nm之间,通过修饰该吸收峰范围可发生变化,具有激发光连续,荧光稳定性好且可调等优点,因此在器件发光领域如发光二极管(light-emitting diode,LED)器件等具有良好的应用前景4-8。此外作为碳族大家庭中的一员,纳米碳点同样具有低毒性和良好的生物相容性,通过修饰可与其他生物分子相互作用,作为脱氧核糖核酸(deoxyribonucleic acid,DNA)等的标记材料,因此在生物医药领域同样具有广阔应用需求9-12。
纳米碳点的制备方法主要有两种13-15,当碳链骨架较大时,把碳骨架上的纳米颗粒剥落下来,制成碳纳米点,称为自上而下法,它主要涵盖电弧放电法、电化学氧化法、激光刻蚀法等。但自上而下法将碳骨架彻底粉碎成纳米颗粒比较困难,产率较低。另外一种是当碳颗粒比较小时,对其进行润饰、钝化而得到碳点,也称自下而上法。主要有燃烧法、超声波法、有机物碳化法、微波法和水热合成法等。自下而上法具有制备方法简便、成本低廉且可大批量生产的优点。
微波法制备纳米碳点是近年来被发展的一种方法16,其制备方法十分便捷,且碳点的粒径和光致发光性质与微波加热时间有关。通常将碳源如葡萄糖等糖类、甘油、有机酸类等与钝化剂配合使用可以实现较好的发光增强效果17-20,但碳源组分的反应机理尚不明确。同时有文献报道pH值对于特定材料组分下纳米碳点的形成具有重要作用21。为此澄清微波法制备纳米碳点的反应机理以及 pH值对于制备纳米碳点性质的影响规律对于高性能纳米碳点的制备及应用具有重要理论价值。本文采用微波法通过控制微波功率、反应时间以及溶液 pH值制备了一系列以葡萄糖为碳源、聚乙二醇400 (polyethylene glycol-400,PEG-400)为钝化剂的纳米碳点。通过测试其激发光谱、发射光谱等对纳米碳点的发光性质进行了表征,并通过紫外吸收光谱与傅里叶红外光谱对参数优化后的纳米碳点进行表面官能团的表征与分析,最终揭示出葡萄糖微波加热的反应机制以及对获得纳米碳点发光性质的影响规律。
2 实验部分
2.1 原料与试剂
葡萄糖,分析纯,国药集团化学试剂有限公司;氢氧化钠,纯度≥ 96.0%,国药集团化学试剂有限公司;盐酸,纯度 36.0%,北京化工厂;聚乙二醇400,分析纯,国药集团化学试剂有限公司;去离子水,自制。
2.2 实验方法
2.2.1 纳米碳点的制备
纳米碳点的制备采用传统的微波合成工艺,具体流程为:将2 g葡萄糖溶于9 mL去离子水中,加入5 mL PEG-400,并采用上海亚荣生化仪器厂的90-磁力搅拌器充分混合并溶解,得到澄清的溶液。随后将该溶液放置于微波炉(格兰仕公司,型号:WP700,最大输出功率700 W)中,调整微波功率与微波时间进行加热,工艺参数详见表1,即得到溶液由无色透明渐至淡黄色或深黄色,即得到纳米碳点,将所得纳米碳点溶液定容至100 mL。其中为对比微波加热对于反应过程中的作用,选取 1号样品为空白样品进行对照实验。其次通过时间和功率调整优化出最佳参数,并利用配好的HCl 溶液(36% (w),1 mol·L-1)和 NaOH 溶液(1 mol·L-1)来调节定容后纳米碳点溶液的pH值,分别调整为3、5、7和9,研究pH值对纳米碳点发光性能的影响规律。
2.2.2 纳米碳点的性质表征
采用英国爱丁堡仪器有限公司F900型荧光光谱仪测试所制备纳米碳点的发射光谱和激发光谱,将少量溶液润洗比色皿之后取少量溶液于比色皿中进行测试。发射光谱采用波长280-380 nm范围内每隔10 nm激发,测试范围至800 nm。而通过将对应每个发射光谱的强度最高值与激发波长作图,即得到试样的激发光谱。
经过发射光谱和激发光谱测试并优化后固化微波功率和时间,进一步针对不同pH值纳米碳点的紫外吸收光谱(ultraviolet (UV) absorptionspectra)和傅里叶变换红外光谱(Fourier transform infrared (FTIR) spectra)进行了表征和对比。其中采用美国Varian公司Cary5000型紫外可见红外分光光度计测紫外吸收谱,可以获得纳米碳点最大吸收波长,而且还可以推测出纳米碳点表面存在的基团,测试范围为200-800 nm。将纳米碳点常温烘干后,采用Varian公司Excalibur 3100型傅里叶红外光谱仪进行了傅里叶红外光谱测试,扫描范围是450-4500 cm-1,扫描次数256次。通过紫外-可见光谱和傅里叶红外光谱分析,分析纳米碳点表面官能团的变化过程,最终揭示pH值对于纳米碳点发光性能的影响规律。
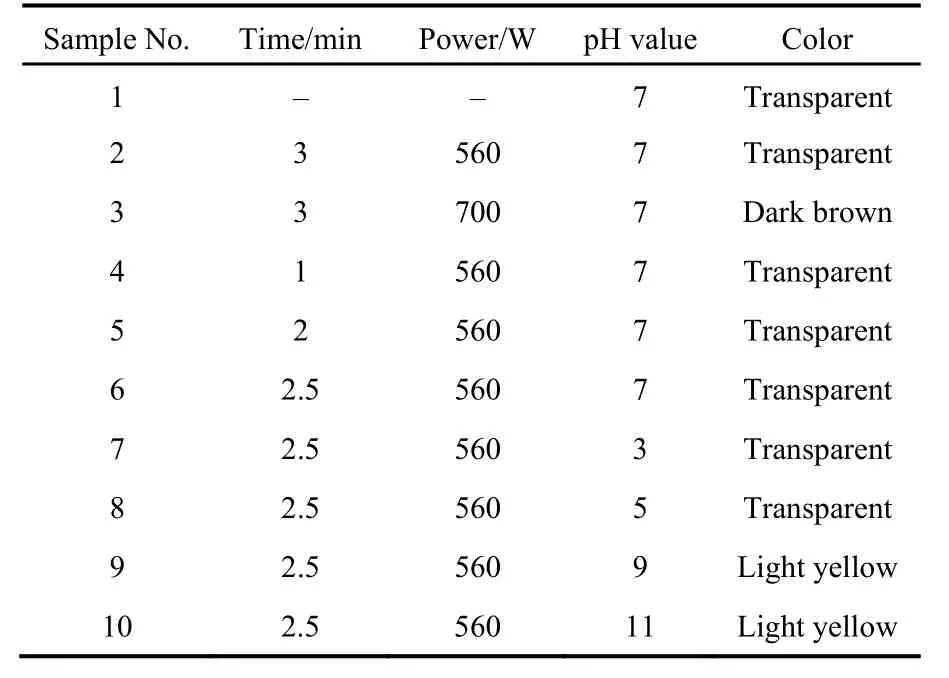
表1 微波合成纳米碳点的工艺参数Table 1 Parameters of nano carbon dots prepared by microwave synthesis.
3 结果与讨论
3.1 微波功率对纳米碳点发光性质的影响
首先探讨了微波功率对纳米碳点发光性质的影响影响,采用微波加热时间3 min,微波加热功率分别为560和700 W。其中低功率加热样品呈现无色透明态,而高功率加热样品呈现深棕色。进一步测试了二者在紫外光(波长 280-380 nm)激发下的发射光谱,示于图1。可以看到采用低功率加热的样品随紫外光波长的增加发光强度先增加后降低,而高功率加热样品在360-575 nm之间只有一个弱的大范围发光区,随波长增加反而降低。进一步作出两种功率加热制备的纳米碳点的激发光谱,见图2。可以看到对于低功率加热的样品在340 nm激发下,呈现出最强的靛色发光(441 nm),而高功率加热样品未表现出明显发光。

图1 560 W (a)和700 W (b)功率下微波加热3 min制备纳米碳点在280-380 nm的发射光谱Fig.1 Emission spectra of nano carbon dots at 280-380 nm with microwave heating for 3 min under 560 W (a) and 700 W (b).
3.2 微波反应时间对纳米碳点发光性质的影响规律

图2 不同功率下微波加热3 min制备纳米碳点在280-380 nm的激发光谱Fig.2 Excitation spectra of nano carbon dots at 280-380 nm with microwave heating for 3 min under different power.

图3 不加热(a)与加热2.5 min (b)制备纳米碳点的发射光谱Fig.3 Emission spectra of nano carbon dots without heating and with heating for 2.5 min.
进一步考察了在微波功率560 W下,微波加热时间从不加热到加热 3 min之间的发射光谱与激发光谱,不加热与加热2.5 min制备纳米碳点的发射光谱图示于图3和图4。从图3(a)中不加热制备的纳米碳点发射光谱可以看出,伴随激发光波长的增加,其发射光谱的最高峰由320 nm向高波长方向移动,但强度逐渐降低,即使是280 nm激发下强度也很低。而随着微波加热时间增加至 1和2 min,其随激发波长增加的发射光谱与不加热下呈现相似的形状和变化趋势,只是发光强度略有不同。随着加热时间增加至2.5 min,纳米碳点的发射光谱呈现出不同的变化规律,见图 3(b)。此时发射光谱的强度明显增加,而且伴随激发波长的增加,发射光谱的峰值略 有移动,当波长370 nm激发下,对应451 nm的蓝光发射最强。进一步将加热3 min的纳米碳点激发光谱一并比较,示于图4中。可以看到只有在加热2.5 min下纳米碳点激发光谱呈现显著的峰值。当加热时间增加至3 min时,此时激发光谱虽也出现峰值,但整体峰值较低,且不够明显。为此优化出560 W下加热2.5 min时纳米碳点的发光性能最佳。
3.3 pH值对纳米碳点发光性质的影响
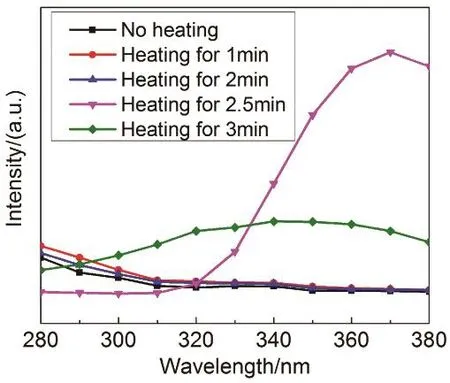
图4 560 W功率下加热不同时间制备纳米碳点的激发光谱Fig.4 Ecitation spectra of nano carbon dots prepared by heating for different time at 560 W.
通过盐酸和氢氧化钠将纳米碳点的 pH值分别调整为3、5、9和11,测试其发射光谱与激发光谱。发现不同pH值下发射光谱的形状与pH值为 7的纳米碳点发射光谱形状较为接近,只是激发光波长与强度略有差异。不同pH值下纳米碳点的激发光谱示于图5。从图5可以看出,随着pH值降低,pH = 3和pH = 5时,纳米碳点在350 nm左右表现出最强的发光峰。相比而言,pH = 5时,发光强度更高。而伴随pH值从中性变为碱性,可见其最强发光峰的激发光波长显著向高波长方向移动,且发光峰强度显著升高。显然,对于pH值为9和11时,最大380 nm波长激发,纳米碳点的发光强度尚未达到最高值。
3.4 纳米碳点发光机理探讨

图5 560 W功率下微波加热2.5 min后不同pH值下纳米碳点的激发光谱Fig.5 Excitation spectra of nano carbon dots prepared by heating at 560 W for 2.5 min under different pH values.
通过以上分析表明,微波加热法制备纳米碳点的过程与微波功率、微波时间及纳米碳点pH值密切相关。且对于微波功率与微波时间而言,均存在最佳值。当反应功率或时间不足时,反应不够充分,发光性质不佳。而当反应功率过高或时间过长后,出现了过反应,此时发光性质同样不佳。
不同 pH值纳米碳点的紫外-可见吸收光谱结果示于图6。其中在700 W下加热试样在紫外与可见光区域表现出很宽的吸收区域,在580 nm可见光黄绿光下吸收系数开始变小,这与其反应过度,溶液呈现棕色有关。而对于560 W加热pH =7的试样,其吸收带范围明显减小,在305 nm处开始降低,基本符合纳米碳点的吸光规律。对于560 W下加热的pH值分别等于5和9的纳米碳点,二者分别在280和275 nm出现显著的吸收峰,这是典型的纳米碳点的紫外吸收峰。该吸收峰与多环芳香族碳氢化合物中 π键有关22。而该吸收峰的移动与碳氢化合物中 π键构型相关,π-π*向高波长方向的红移与扩展共轭效应密切相关23。
进一步通过傅里叶红外光谱分析纳米碳点的表面官能团,可以看到经微波加热反应的纳米碳点,均不同程度的表现出C―O (1092 cm-1)、C=O(1719 cm-1)、C―H (2800-3000 cm-1与 940 cm-1)、OH (3000-3700 cm-1)等键合,见图7所示。当微波反应加热后,560 W加热pH = 7的试样具有较弱的C―O键,但具有较强的C=O键,以及出现C=C键(1643 cm-1),与紫外吸收光谱中π键出现一致。伴随在更高功率下加热,此时C=O键与C=C键均呈现减弱的趋势。相比而言,对于呈现酸性与碱性的样品,C=C双键的强度没有减弱,而C=O双键的相对强度明显减弱。酸性与碱性环境的纳米碳点的官能团差异主要在于C=O键与C=C键的相对含量。
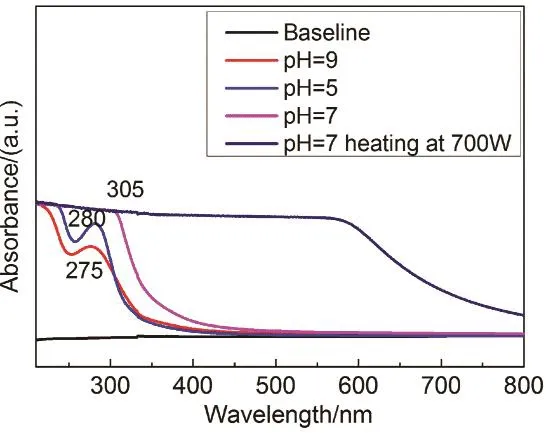
图6 不同pH值纳米碳点的紫外吸收光谱Fig.6 UV absorption spectra of nano carbon dots with different pH values.
对于葡萄糖而言,其自身结构中存在C―O、OH与C―H键。由于葡萄糖与水分子均为极性分子,随着微波能量的输入,作用于分子产生加热作用,待达到所需反应活性时,葡萄糖将逐步发生官能团反应。反应生成了C=O与C=C键,并且具有芳香族特征,表明葡萄糖单体发生了交联。葡萄糖具有诸多的 OH键,伴随微波加热过程很容易发生脱水反应,因此不同单体之间很快便会通过单体间脱水发生聚合反应形成一定聚合度的多糖。随着微波能量的不断积累,不同聚合度的多糖将发生显著的碳化过程,形成芳香族碳氢化合物,从反应产物的官能团来看,这一过程类似于诸多其他碳氢化合物以及水热法中碳化产物的转变过程24。
反应过程中,由于葡萄糖多聚体内仍然存有较多OH键,这些OH键在加热升温过程中可能发生如下反应,首先羟基间脱水,使得其中一羟基中O与同一碳原子形成酮基,如图8中第1和第 2步所示。生成酮基后的多聚体有可能会发生酮基与临近碳原子上的H连接,形成C=C同时保留 OH键,这也是形成芳香族碳氢化合物的基本反应25,如图8中第3步所示。而C=C键也可以通过 OH与临近 H直接脱水,直接形成 C=C双键,但是这个反应要在更高温度进行,同时可能伴随多聚体的竞争重组过程。因此对于增加微波加热功率而言,在 C=O键减少的同时,多聚体的竞争重组占据优势,因此出现 C=C键也减少的现象。
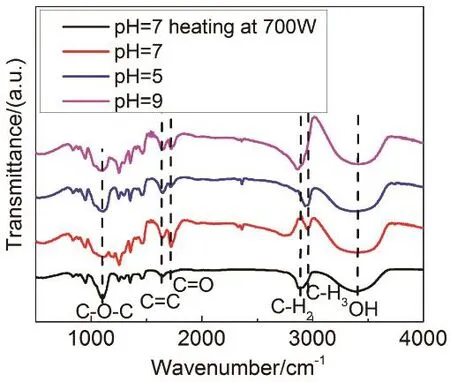
图7 不同pH值纳米碳点的傅里叶变换红外光谱Fig.7 FTIR spectra of nano carbon dots with different pH values.

图8 葡萄糖单体加热反应过程中的键合变化过程示意图Fig.8 Schematic diagram of bonding change of glucose in the heating process.
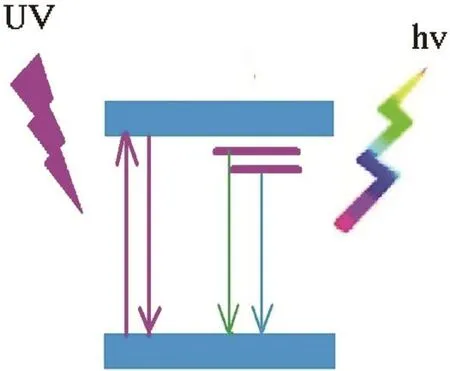
图9 微波法制备纳米碳点的发光机理Fig.9 Luminescence mechanism of nano carbon dots prepared by microwave synthesis.
通过pH值的调整,纳米碳点中C=C键含量变化不明显,但C=O键含量从pH = 7的中性状态,均出现了降低,其中酸性环境会消耗碳点中残留的 OH基团,使得生成酮基的反应向逆反应方向进行。而碱性环境则维持C=C键与C=O键几乎维持相近的程度。对于纳米碳点而言,首先决定其发光特性的因素是尺度。伴随延长反应时间与增加功率,均会使得葡萄糖反应向碳化方向发生,生成具有 C=C键特征的芳香族化合物。以单个芳香族单元苯环为例,尺寸约0.5 nm左右,对于数纳米尺度的碳点而言,10个左右芳香族基本单元是实现纳米碳点最佳发光的结构。随着时间或功率显著增加,进一步的碳化将使得碳粒子不再具有发光特性。其次则是交联碳化过程中的C=C键与C=O键的共同作用。特定化学键合的形成伴随表面能量陷阱的产生,受到pH值的微观调控。中性环境下,C=O键多于C=C键;酸性环境下C=O键少于C=C键;碱性环境下C=O键与 C=C键基本持平。键合的类型与比例也直接决定了纳米碳点的光学带隙宽度及激子束缚能,从而产生了吸收带隙的差异以及发光强度与发光波段的对应性。如图 9所示,为微波法合成纳米碳点的发光原理图,当特定波长的紫外光照射纳米碳点时,此时能量满足光学带隙的光子将发生跃迁,跃迁后的光子特别是在邻近的表面缺陷陷阱聚集,通过返回到基态进而发射出不同波长的可见光。而该表面缺陷陷阱则是由纳米碳点的骨架(尺度与 C=C键)、表面的各种官能团(C―O―C键、OH键、特别是C=O键等)共同作用实现的。对于酸性和碱性下纳米碳点的光学带隙可对应紫外吸收光谱中的吸收峰,中性下对应吸收截止限。而缺陷能级则与激发与发射光谱相关,相比而言,碱性环境下纳米碳点的缺陷能级向高波长方向移动,其激子束缚能相对较小。
4 结 论
采用微波加热法合成纳米碳点,当微波功率为560 W,反应时间为2.5 min时,获得纳米碳点发光性能最佳。当波长370 nm激发下,对应451 nm的蓝光发射最强。伴随溶液pH值从酸性变为碱性,纳米碳点最强发光峰的激发光波长由 350 nm显著向高波长方向移动,且发光峰强度显著升高。紫外吸收光谱与傅立叶红外光谱显示反应过程中形成了多环芳香族碳氢化合物,表明微波加热过程中是单糖像多糖聚合并最终发生碳化的过程。不同pH值下的纳米碳点,伴随对于纳米碳点中C=C键与C=O键比例的调整,从而实现对纳米碳点的光学带隙宽度及激子束缚能等的综合调控,使得纳米碳点呈现出不同的发光规律。本文获得的研究结果为纳米碳点的结构控制与发光性能优化提供了理论基础。
(1) Xu, X. Y.; Ray, R.; Gu, Y. L.; Ploehn, H. J.; Gearheart, L.; Raker, K.;Scrivens, W. A. J. Am. Chem. Soc. 2004, 126 (40), 12736.doi: 10.1021/ja040082h
(2) Cao, L.; Wang, X.; Meziani M. J.; Lu, F.; Wang, H.; Luo, P. G.; Lin,Y.; Harruff, B. A.; Veca, L. M.; Murray, D.; Xie, S. Y.; Sun, Y. P.J. Am. Chem. Soc. 2007, 129 (37), 11318.doi: 10.1021/ja073527l
(3) Feng, C.; Deng, X. Y.; Ni, X. X.; Li, W. B. Acta Phys. -Chim. Sin.2015, 31 (12), 2349. [冯 昌, 邓晓燕, 倪晓晓, 李卫兵.物理化学学报, 2015, 31 (12), 2349.]doi: 10.3866PKU.WHU.WHXB- 20150281
(4) Guo, X.; Wang, C. F.; Yu, Z. Y.; Chen, L.; Chen, S. Chem. Commun.2012, 48 (21), 2692.doi: 10.1039/C2CC17769B
(5) Wang, F.; Chen, Y. H.; Liu, C. Y.; Ma, D. G. Chem. Commun. 2011,47 (12), 3502. doi: 10.1039/C0CC05391K
(6) Sun, Y. P.; Zhou, B.; Lin, Y.; Fernando, K. A. S.; Pathak, P.; Meziani,M. J.; Harruff, B. A.; Wang, X.; Wang, H.; Luo, P. G.; Yang, H.;Kows, M. E.; Chen, B.; Veca, L. M.; Xie, S. Y. J. Am. Chem. Soc.2006, 128 (24), 7756. doi: 10.1021/ja062677d
(7) Yang, S. T.; Cao, Li.; Luo, P. G.; Lu, F.; Wang, X.; Wang, H.;Meziani, M. J.; Liu, Y.; Qi, G.; Sun, Y. P. J. Am. Chem. Soc. 2009,131 (32), 11308. doi: 10.1021/ja904843x
(8) Zhu, S.; Meng, Q.; Wang, L.; Zhang, J.; Song, Y.; Jin, H.; Zhang, K.;Sun, H.; Wang, H.; Yang, B. Angew. Chem. Int. Edit. 2013, 52 (14),3953. doi: 10.1002/anie,. 201300519
(9) Huang, P.; Lin, J.; Wang, X. S.; Wang, Z.; Zhang, C. L.; He, M.;Wang, K.; Chen, F.; Li, Z. M.; Shen, G. X.; Cui, D. X.; Cui, D. X.;Chen, X. Y. Adv. Mater. 2012, 24 (37), 5104.doi: 10.1002/adma.201200650
(10) Lai, C. W.; Hsiao, Y. H.; Peng, Y. K.; Chou, P. T. J. Mater. Chem.2012, 22 (29), 14403. doi: 10.1039/C2JM32206D
(11) da Silva, J. C. G. E.; Goncalves, H. M. R. Trac-Trend. Anal. Chem.2011, 30 (8), 1327. doi: 10.1016/j.trac. 2011.04.009
(12) Liu, C. J; Zhang, P.; Zhai, X.; Tian, F.; Li, W.; Yang, J.; Liu, Y.;Wang, H.; Wang, W.; Liu, W. Biamaterials 2012, 33 (13), 3604.doi: 10.1016/ j.biomaterials.2012.01.052
(13) Bao, L.; Zhang, Z. L.; Tian, Z. Q.; Zhang, L.; Liu, C.; Lin, Y.; Qi, B.;Pang, D. W. Adv. Mater. 2011, 23 (48), 5801.doi: 10.1002/adma.201102866
(14) Yan, F. Y.; Zhou, Y.; Wang, M.; Dai, L. F.; Zhou, X. G.; Chen, L.Prog. Chem. 2014, 26 (1), 61. [颜范勇, 邹 宇, 王 猛, 代林枫,周旭光, 陈 莉. 化学进展, 2014, 26 (1), 61.]doi: 10.7536/PC130643
(15) Wang, S. D.; Zhu, Z. F.; Chang, Y. J.; Wang, H.; Yuan, N.; Li, G. P.;Yu, D. B.; Jiang, Y. Nanotecnology 2016, 27 (29), 295202.doi: 10.1088/0957-4484/29/295202
(16) Zhu, H.; Wang, X.; Li, Y.; Wang, Z.; Yang, F.; Yang, X. Chem.Commun. 2009, No. 34, 5118. doi: 10.1039/B907612C
(17) Jaiswal, A.; Ghosh, S. S.; Chattopahyay, A. Chem. Commun. 2012,48 (3), 407. doi: 10.1039/C1CC15988G
(18) Chandra, S.; Das, P.; Bag, S.; Laha, D.; Pramanik, P. Nanoscale 2011,3, 1533. doi: 10.1039/C0NR00735H
(19) Wang, K.; Gao, Z. C.; Gao, G.; Wo, Y.; Wang, Y. X.; Shen, G. X.;Cui, D. X. Nanoscale. Res. Lett. 2013, 8, 122.doi: 10.1186/1556-276X-8-122
(20) Wang, X. H.; Qu, K. G.; Xu, B. L.; Ren, J. S.; Qu, X. G. J. Mater.Chem. 2011, 21, 2445. doi: 10.1039/C0JM02963G
(21) Jiang, J.; He, Y.; Li, S. Y.; Cui, H. Chem. Commun. 2012, 48, 9634.doi: 10.1039/C2CC34612E
(22) Chitrea, S. A.; Loboa, G. M.; Rathoda, S. M.; Smitha, R.;Livingstoneb, C. J. Chromatogr. B 2008, 864 (1), 173.doi: 10.1016/j.jchromb.2008.01.040
(23) Liu, Y.; Chu, Y.; Yang, L.K. Mater. Chem. Phys. 2006, 98 (2-3), 304.doi: 10.1016/j.matchemphys.2005.09.025
(24) Li, H. T.; He, X. D.; Liu, Y.; Huang, H.; Lian, S. Y.; Lee, S. T.; Kang,Z. H. Carbon 2011, 49, 605.doi: 10.1016/j.carbon.2010.10.004
(25) Tang, M.M.; Bacon, R. Carbon 1964, 2, 211.doi: 10.1016/00086223
