S掺杂促进Fe/N/C催化剂氧还原活性的实验与理论研究
2018-01-10周志有张新胜孙世刚
陈 驰 张 雪 周志有 张新胜 孙世刚,
S掺杂促进Fe/N/C催化剂氧还原活性的实验与理论研究
陈 驰1,2张 雪2周志有2张新胜1,*孙世刚1,2,*
(1华东理工大学化工学院,化学工程联合国家重点实验室,上海 200237;2厦门大学化学化工学院,固体表面物理化学国家重点实验室,厦门 361005)
向Fe/N/C非贵金属催化剂中再引入S掺杂是进一步提高其氧还原催化活性的有效方法。为了探究活性提高的原因,本文以三聚氰胺-甲醛树脂为前驱体,氯化钙为模板,氯化铁为铁源,通过添加硫氰化钾(KSCN)来控制热解催化剂的S掺杂量。通过对比分析催化剂的物化性质,结合密度泛函理论(DFT)计算,分析S掺杂促进Fe/N/C催化剂氧还原活性的原因。透射电子显微镜(TEM)和N2吸脱附等温线测试结果表明,S元素可抑制含铁纳米粒子的形成,促使形成多孔碳结构,提高比表面积。X射线光电子能谱(XPS)结果表明,适量S前驱体可实现较高的S掺杂含量,得到最优的活性,过量的S反而会导致Fe和S的掺杂量同时降低,影响活性。DFT计算结果表明在Fe-N4大环中引入S掺杂,可增强O2分子和中间体OOH与Fe-N4结构中的Fe的相互作用,促进形成Fe―O键,从而导致O―O键的键能显著降低,为后续反应O―O键的断裂提供可能,促进ORR反应的进行。
氧还原反应;非贵金属催化剂;Fe/N/C材料;S掺杂;密度泛函理论
1 引 言
质子交换膜燃料电池(PEMFC)可将化学能直接转化为电能,不受卡诺循环的限制,因其转化效率高、清洁无污染等优点备受关注1。催化剂是燃料电池的核心部件,其主要活性组分为铂。但是铂的价格昂贵、储量稀少、易中毒失活,导致燃料电池成本居高不下,使用寿命无法满足实际应用需求2。燃料电池阴极氧还原反应(ORR)的动力学十分缓慢,阴极所需的铂催化剂用量数倍于阳极3。因此,开发资源储量丰富、高性能、低成本的非贵金属氧还原催化剂对于降低燃料电池的成本,促进燃料电池大规模应用具有重要意义4。
近十年来,过渡金属(Fe、Co)掺杂的M/N/C氧还原催化剂的性能已显著提升,展现出良好的应用潜力5–7。研究表明,原始的碳材料对氧还原的催化活性非常低,如石墨烯、碳纳米管、碳黑等8。选用电负性、原子尺寸等性质与碳原子不同的杂原子 (如N、S、B、P等)进行掺杂,可有效的调控碳材料的表面电子结构,引入电荷缺陷和结构缺陷,促进氧气吸附和电荷转移9–11。两种杂原子共掺杂还可引入协同效应,进一步提高氧还原催化活性12。但在酸性介质中,过渡金属通常是活性中心不可或缺的组成部分13。Zelenay等人14以聚苯胺包覆碳黑颗粒,添加Fe和Co的无机盐,制备了一系列具有高ORR活性的PANI-M-C催化剂。我们也以高含氮量的聚间苯二胺包裹的炭黑为碳、氮前驱体15,并优化了铁源,如采用硫氰化铁(Fe(SCN)3)配合物,在引入Fe的同时也引入S元素16,大幅提升了催化剂的ORR活性。由于碳载体相同,S掺杂对于催化剂的形貌结构没有明显影响,仅比表面积稍有提高。Ferrandon等人17的研究表明,S元素的添加可抑制热处理过程中形成碳化铁,有利于形成Fe-N4活性中心。尽管如此,对于S掺杂促进Fe/N/C催化剂氧还原活性的原因仍不太明确。DFT理论计算结果将石墨烯中N-S二元共掺杂引入的协同效应归因于N协同下的电荷密度变化,以及S原子可极化的轨道增强周围原子的电子极化18。但该模型没有考虑过渡金属对于活性中心的作用,因此可能不适用于酸性条件下的氧还原反应。近来更多的模型研究倾向于支持嵌入在碳平面内的Fe-N4成分为活性中心的结论,因为这种结构更有利于O2分子的吸附及随后的O2质子化和O―O键的解离19,20。我们通过在单层石墨烯上制备单原子层的Fe/N/C模型催化剂,表明ORR活性与N-Fe物种含量有较好的线性关系21。Zitolo等人22通过扩展X射线吸收精细结构(EXAFS)和X射线吸收近边结构(XANES)技术在不含晶相铁的Fe-N-C催化剂中检测到类卟啉结构的FeN4C12物种,表明ORR的活性中心很可能是FeN4结构。
在本工作中,我们不额外添加碳载体,仅以三聚氰胺-甲醛树脂为碳、氮前驱体,以氯化钙为模板,在铁含量不变的情况下,改变硫氰化钾(KSCN)的添加量,研究S元素对Fe/N/C催化剂的影响。通过对比所制备的催化剂(Resin-FeNS/C)的形貌结构、比表面积、组分含量等性质,发现适量S元素可抑制含铁纳米粒子的形成,促进形成多孔碳,提高比表面积,增强传质;同时引入S掺杂,提高杂原子掺杂含量,其氧还原活性为不含S样品的2.2倍。但过量的S元素反而会降低催化剂中的Fe和S掺杂量,对活性不利。DFT计算以Fe-N4结构为活性中心模型。结果表明,S原子掺杂可增强O2及中间体OOH与中心Fe原子的相互作用,形成Fe―O键,使O―O键键能降低,更容易断裂,从而促进ORR反应的进行。
2 实验部分
2.1 Resin-FeNS/C催化剂的制备
Resin-FeNS/C催化剂的合成过程参照我们已报道的工作23,主要包括前驱体制备和高温热处理两步,制备方法如图1所示,简述如下:
(1) 三聚氰胺-甲醛树脂的制备:将15.6 mmol三聚氰胺粉末和3 mL 37%的甲醛溶液分散于30 mL水中,加入1 mL 0.1 mol∙L−1NaOH溶液,调节溶液的pH值为9–10。70 °C下水浴恒温搅拌1 h后,加入0.2 mL 1 mol∙L−1HCl溶液,调节溶液的pH值为5–6,以加快树脂聚合速度。待溶液出现白色浑浊后,加入20 mmol氯化钙(CaCl2) 粉末和10 mL FeCl3水溶液(1 mol∙L−1,10 mmol),分别作为模板和铁源。以Fe3+: SCN−的摩尔比为1 : 0,1 : 3和1 : 6的比例,分别添加0,30和60 mmol硫氰化钾(KSCN)。升温至85 °C,继续搅拌加热24 h,使树脂充分聚合。之后在85 °C下搅拌加热蒸干溶剂,80 °C下真空干燥过夜,所得固体研磨成粉备用。

图1 Resin-FeNS/C催化剂的合成示意图
(2) 高温热处理:取3 g前驱体于瓷舟中,在Ar气保护下900 °C高温热处理1 h,使树脂碳化,升温速率为5 °C·min−1。冷却至室温后,将样品分散于100 mL 1 mol∙L−1HCl溶液中,80 °C下酸处理7 h。离心分离收集固体,洗涤3次后,样品在80 °C下真空干燥3 h。所得固体在Ar气保护下,900 °C二次高温热处理3 h,冷却至室温后,所得催化剂标记为Resin-FeNS/C-/,其中/为Fe3+和SCN−的摩尔比,分别为1/0,1/3和1/6。
2.2 催化剂的物化性质表征
X射线光电子能谱(XPS)采用Qtac-100 LEISS-XPS仪测试(ION TOF公司,德国),分析催化剂的表面组分含量及其化学态;采用日本Rigaku公司的Ultima IV型X射线衍射仪(XRD)测试催化剂的晶相结构,测试角范围为10°–90°,步长为10 (°)∙min−1;采用日本电子株式会社的JEM-1400透射电子显微镜(TEM)观察催化剂的形貌结构,加速电压为100 kV;采用美国Micromeritics公司的TriStar II 3020仪测定催化剂的氮气吸脱附等温线及比表面积。
2.3 电化学性能测试
电化学测试采用传统的三电极体系,30 °C下水浴恒温。测试在美国Pine公司产的旋转圆盘电极(RDE)系统上进行,采用CHI 760D电化学工作站。玻碳工作电极在使用前依次用1 μm和0.3 μm的Al2O3粉末抛光、超声清洗三次,玻碳盘直径为5 mm。将6 mg催化剂与0.5 mL水、0.45 mL乙醇和0.05 mL 5% Nafion混合,超声1 h充分分散,制得催化剂浆料。用移液枪移取20 μL 催化剂浆料滴涂在玻碳盘上,红外灯烤干,制得工作电极,催化剂的载量为0.6 mg·cm−2。对电极为1 cm × 10 cm的石墨片;参比电极为自制的可逆氢电极(RHE)。电解质溶液为氧气饱和的0.1 mol∙L−1H2SO4溶液;电极转速为900 r·min−1;电位扫速为10 mV·s−1。在氮气饱和的0.1 mol∙L−1H2SO4溶液中以相同的电位区间和扫速记录催化剂的双电层背景曲线,将氧气饱和时记录的曲线扣除双层电容后即为催化剂的氧还原极化曲线。
通过Koutecky-Levich方程可校正扩散控制的影响,计算得到催化剂的动力电流k:
Koutecky-Levich方程: 1/= 1/L+ 1/K(1)
式中为表观电流,L为极限扩散电流,K为动力电流。
质量活性m可通过将k相对于电极表面的催化剂载量cat进行归一化得到:

质量活性:jm = ik/Mcat(2)
2.4 计算方法和模型
采用基于密度泛函理论(DFT)的CP2K程序包24对FeNC结构中S掺杂的所有可能位置及其吸附体系进行结构优化。采用混合高斯和平面波双重基组方法(GPW)可以在较低的波函数截断能下获得较好的能量和力的计算精度,而价电子和包含内层电子的原子核之间的相互作用则使用Geodecker-Teter-Hutter (GTH)赝势来描述25–27。其中,交换关联部分主要采用杂化泛函 B3LYP,基组用TZV2P-MOLOPT-GTH基组,赝势用GTH-BLYP,截断能为500 eV,点选择2 × 2 × 1。
选用(6 × 6)的石墨烯模型,在其中嵌入一个Fe-N4中心,形成具有66个碳原子,4个N原子,1个Fe原子的Fe-N4/C结构。为避免Fe-N4/C层间的干扰,真空层取为15 Å。能量收敛精度为10−5eV。计算中加入自旋。
吸附能ad的计算按照以下公式:

Ead = EM* − (E* + EM)(3)
其中E为掺杂并吸附分子后的石墨烯体系能量,*为掺杂后的石墨烯体系能量,E为分子的能量。
吸附的O2分子第一步加H生成OOH的自由能能变是以吸附的O2的能量为基准,以标准氢电极(SHE)为参比电极,以1/2 H2的能量代替H+和−的能量条件下计算得到。

图2 Resin-FeNS/C-a/b (a/b = 1/0,1/3,1/6)催化剂的TEM图
(a) Resin-FeNS/C-1/0, (b) Resin-FeNS/C-1/3, (c) Resin-FeNS/C-1/6.
3 结果与讨论
图2为前驱体中不同Fe3+: SCN−摩尔比的催化剂的TEM图。由图可见,Resin-FeNS/C-/(/= 1/0,1/3,1/6)三者的形貌结构存在明显差异。当前驱体中不含S元素时(/= 1/0),催化剂中同时存在两种结构,即多孔碳和包裹着Fe纳米粒子的碳纳米管,即类似于之前文献报道的豆荚铁碳纳米管28,如图2(a)所示。当前驱体中存在S元素时(/= 1/3和1/6),则催化剂中都仅可观察到多孔碳结构,并且二者的形貌结构非常类似,如图2(b)、(c)所示。豆荚铁碳纳米管的形成可能是由于在高温下Fe离子被还原成单质Fe纳米粒子,其催化有机物分解形成碳纳米管并包覆在表面。类似条件下也可能会形成碳层包覆的Fe3C纳米粒子结构29。而S元素在高温下则会与Fe反应生成FeS,抑制单质Fe纳米粒子的形成,并在酸处理过程中被去除,因此不会形成豆荚铁结构。另一方面,前驱体中添加的CaCl2粉末均匀分散在三聚氰胺-甲醛树脂中,可防止高温下树脂交联形成块状碳,同时起到造孔模板的作用,促进形成多孔碳结构,有利于提高比表面积,促进传质。CaCl2模板和FeS颗粒均可在酸处理过程中完全去除,因此,在图2(b)、(c)中没有观察到纳米颗粒的物质。
通过XRD技术表征Resin-FeNS/C-/(/= 1/0,1/3,1/6)催化剂的晶相物种。如图3所示,三个催化剂的XRD谱图在26°位置均出现一个宽峰,对应于石墨烯(002)特征峰,其源于催化剂中的多孔碳或碳纳米管结构。而Resin-FeNS/C-1/0在44.3°位置还出现一个较强的峰,可归属于Fe纳米粒子的衍射峰,该结果与TEM观察到的结果一致。除此之外,在Resin-FeNS/C-/(/= 1/3,1/6) 两个催化剂的XRD谱图中都未明显出现其它晶相物种衍射峰,也表明CaCl2模板和其它非活性物种在酸处理过程中已被去除。
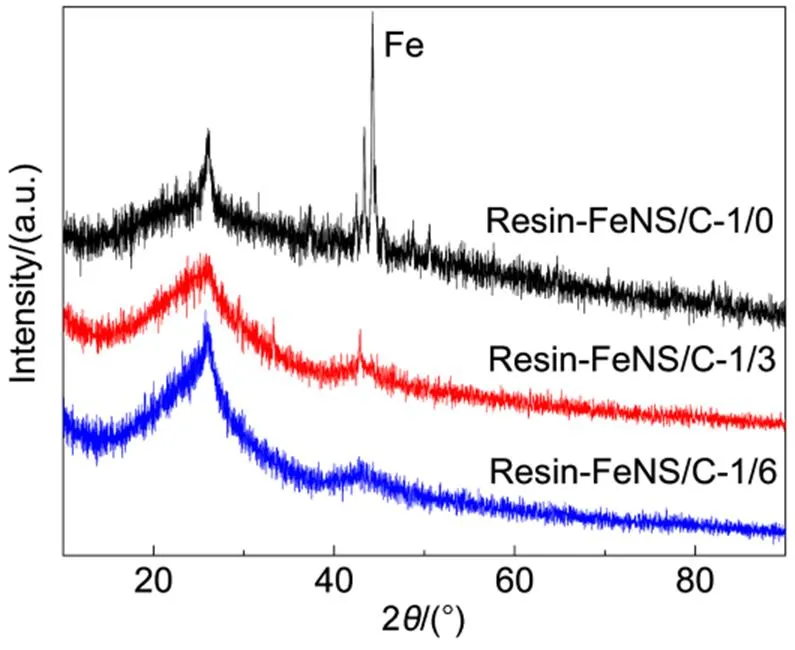
图3 Resin-FeNS/C-a/b (a/b = 1/0, 1/3, 1/6)催化剂的XRD谱图

图4 Resin-FeNS/C-a/b (a/b = 1/0,1/3,1/6)催化剂的氮气吸脱附等温线
(1) Resin-FeNS/C-1/0, (2) Resin-FeNS/C-1/3, (3) Resin-FeNS/C-1/6.

图5 Resin-FeNS/C-a/b (a/b = 1/0,1/3,1/6) 催化剂的XPS全谱分析(a)和N 1s (b), S 2p (c), Fe 2p (d)的高分辨XPS谱图
(1) Resin-FeNS/C-1/0, (2) Resin-FeNS/C-1/3, (3) Resin-FeNS/C-1/6.
催化剂的形貌结构与表面性质有密切关系,通过测试氮气吸脱附等温线可以表征催化剂的比表面积和孔隙信息,如图4所示。由于Resin-FeNS/C-1/0中存在豆荚铁结构,增大了催化剂的密度,因此其比表面积也较小,仅为536 m2·g−1。Resin-FeNS/C-/(/= 1/3,1/6) 中都仅含多孔碳结构,因此二者的比表面积非常接近,分别为775和793 m2·g−1,明显高于Resin-FeNS/C-1/0。另外,在蒸汽压接近零的低压区,催化剂对N2的吸附量 (主要是微孔吸附) 很低,且吸附量随相对压强的增大而逐渐升高,表明催化剂是以外表面积为主,这也与其多孔碳的结构一致。
采用XPS分析Resin-FeNS/C-/(/= 1/0,1/3,1/6)催化剂的表面元素组成。图5分别显示了三个催化剂的XPS宽谱,以及N 1、S 2和Fe 2的高分辨XPS谱图。由图可见,Resin-FeNS/C-/的主要组成元素为C、N、O、S和Fe。三个催化剂中的元素相对原子含量如表1所示。催化剂的N和C都来源于三聚氰胺-甲醛树脂,因此在同样的热处理条件下,三者的N含量都较高且比较接近。当添加SCN−引入S元素的同时也会引入部分N源,因此Resin-FeNS/C-/(/= 1/3,1/6) 中的N含量比Resin-FeNS/C-1/0稍高,并且Resin-FeNS/C-1/6的含N量也稍高于Resin-FeNS/C-1/3,如图5(b)所示。通过图5(c)、(d)的对比可以看出,在前驱体中含有过多SCN−离子的情况下,Resin-FeNS/C-1/6中的S含量和Fe含量却都低于Resin-FeNS/C-1/3。显然,添加适量的S前驱体可与过量的Fe元素形成FeS,避免形成豆荚铁结构,并且引入S元素掺杂。但是若前驱体中的S元素过量,则会竞争形成更多的FeS,并在酸处理过程中被去除,导致掺杂进催化剂中的S元素和Fe元素含量同时降低。另外,在不添加S元素的Resin-FeNS/C-1/0中也可检测到微量的S,这可能是在热处理过程中引入的杂质。由图5(d)可见,虽然在Resin-FeNS/C-1/3中不含Fe纳米粒子,但是其Fe含量却比Resin-FeNS/C-1/0略高,这是因为XPS是表面探测技术,无法检测到碳纳米管内部的Fe纳米粒子的信号。进一步通过CHNS元素分析表征催化剂中的N、S含量,确认Resin-FeNS/C-1/6中的S元素含量仅为Resin-FeNS/C-1/3的一半左右,其结果与XPS分析的结果一致,如表2所示。
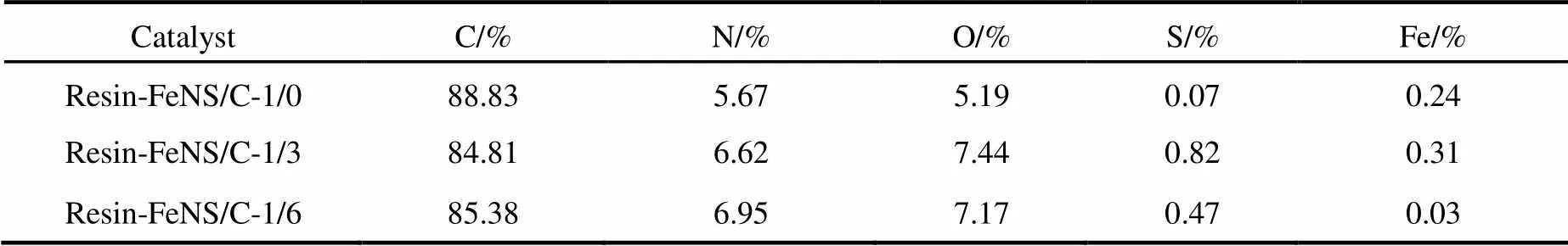
表1 XPS谱图分析的Resin-FeNS/C-a/b (a/b = 1/0,1/3,1/6)催化剂中各元素的相对原子含量

表2 Resin-FeNS/C-a/b (a/b = 1/0,1/3,1/6) 催化剂的CHNS元素分析
通过旋转圆盘电极技术对Resin-FeNS/C-/(/= 1/0,1/3,1/6) 催化剂的氧还原活性进行测试。图6展示了三个催化剂在氧气饱和的0.1 mol∙L−1H2SO4溶液中的ORR极化曲线和活性对比。由图可见,活性最高的Resin-FeNS/C-1/3的半波电位可达到0.809 V,该数值分别比Resin-FeNS/C-1/0和Resin-FeNS/C-1/6的半波电位高22 mV和12 mV。在0.8 V电位下的表观电流密度为2.38 mA·cm−2,经Koutecky-Levich方程校正传质扩散的影响,可得到该电位下的动力电流,进一步与催化剂载量归一化,可知Resin-FeNS/C-1/3在0.8 V电位下的质量活性可达到9.8 A·g−1。该质量活性分别为Resin-FeNS/C-1/0 (4.4 A·g−1)和Resin-FeNS/C-1/6 (6.4 A·g−1)的2.2倍和1.5倍,如图6(b)所示。综合上述物化性质表征和电化学测试可以看出,在Fe/N/C催化剂中额外掺杂S可有效提高ORR催化活性,其原因可能包含以下几点:(1) 避免形成含Fe纳米粒子,提高了催化剂的比表面积,形成多孔结构促进传质,提高活性位点利用率;(2) S原子因其特有的电负性、电子极性和较大的原子尺寸,可引入更多电子缺陷和结构缺陷,促进氧气吸附30;(3) S、N共掺杂引入协同效应,进一步提高ORR活性31。另一方面,前驱体中S元素的含量需仔细优化,以得到最优活性。在催化剂的形貌结构与比表面积相近的条件下,Resin-FeNS/C-1/6的活性较低则是由于过量的S使催化剂中的S和Fe掺杂量同时降低,影响活性。相比于比表面积等性质的提高,S掺杂引入的活性中心电子结构的变化对促进ORR活性起更主要的作用。
通过密度泛函理论计算分析S原子掺杂促进Fe/N/C催化剂ORR活性的原因。如前言所述,模型催化剂的研究表明活性中心可能为嵌入在碳平面内的Fe-N4结构,因此我们以镶嵌Fe-N4结构的石墨烯平面为活性中心模型,将周围的其中1个C原子替换成S原子,构造出S掺杂的Fe-N4/C模型结构,并认为O2及反应中间物种都吸附于Fe原子上。通常情况下,电负性的差异引起的电子得失会导致电荷密度重新分布,因此掺杂原子的位置越靠近活性中心,对活性的影响越大。我们考虑了以下4种可能位置,分别标记为Fe-N4S1/C、Fe-N4S2/C、Fe-N4S3/C和Fe-N4S4/C,如图7(a)所示。Fe-N4S1/C、Fe-N4S2/C和Fe-N4S3/C三种模型中的S原子掺杂在Fe-N4中心环内,而Fe-N4S4/C中的S原子掺杂在Fe-N4中心环外。对以上模型结构进行优化后,由图7(b)可见,所有Fe-N4/C结构在掺杂S之后,S原子突出石墨烯平面形成一个四面体结构,同时Fe-N4/C的平面结构发生弯曲。通过计算可知,这四种掺杂结构的能量依次分别为−543.743 eV、−543.756 eV、−543.748 eV和−543.738 eV,相近的能量表明这四种构型的稳定性没有明显差异。

图6 Resin-FeNS/C-a/b (a/b = 1/0,1/3,1/6)催化剂的氧还原极化曲线(a)和质量活性与半波电位的对比(b)
Catalyst loadings were 0.6 mg·cm−2; The electrolyte was O2-saturated 0.1 mol∙L−1H2SO4; Rotating speed was 900 r·min−1; Scan rate was 10 mV∙s−1.

图7 Fe-N4/C上四种不同掺S位置(a)及优化后的S掺杂Fe-N4/C的侧视图(b)
燃料电池阴极上发生的氧还原反应可通过4电子过程将O2直接还原生成H2O (O2+ 4H++ 4−→ 2H2O),相比2电子过程反应生成H2O2(O2+ 2H++ 2−→ H2O2) 具有更高的转化效率,并可避免腐蚀性的H2O2对燃料电池寿命的影响。通常认为4电子转移路径如下32,33:

O2 + * → O2*(4) O2* + H+ + e− → OOH*(5) OOH* + H+ + e− → O* + H2O(6) O* + H2O + H+ + e− → OH* + H2O(7) OH* + H2O + H+ + e− → * + 2H2O(8)
其中*,O*,O2*,OH*,OOH* 分别表示催化剂表面以及表面上吸附的O、O2、OH和OOH。在4电子反应过程中,第一个电子转移通常被认为是ORR过程的决速步骤34,而O―O键的断裂是反应能够发生的前提,因此O2或OOH的吸附能太低或太高都不利于ORR反应的进行。O―O键断裂后,吸附的O和OH与溶液中的H+结合生成H2O。
表3和表4给出了反应过程中,Fe-N4/C及S掺杂Fe-N4/C结构(标记为Fe-N4S/C) 吸附不同中间体形成Fe-N4S/C-O2,Fe-N4S/C-OOH,Fe-N4S/C-O和Fe-N4S/C-OH的吸附能和原子间距。当O2吸附在Fe-N4S/C上后,其中一个氧原子垂直于Fe-N4表面,Fe原子被拉出Fe-N4平面,如图8(a)所示。Fe-N4S1/C,Fe-N4S2/C和Fe-N4S3/C上O2的吸附能明显增强(~0.2 eV),Fe―O键长比吸附在Fe-N4/C结构中的短。同时,O―O键的键长相比于孤立状态的O2分子(0.123 nm) 也发生了明显的变化。吸附在Fe-N4/C结构上的O2的O―O键长为0.129 nm,而吸附在Fe-N4S/C结构中的O―O键长分别为0.137 nm (Fe-N4S1/C),0.135 nm (Fe-N4S2/C),0.135 nm (Fe-N4S3/C),0.130 nm (Fe-N4S4/C)。Fe―O键的缩短和O―O键的拉伸有利于O2进行下一步反应。对于S掺杂在Fe-N4环外的Fe-N4S4/C结构,O2的吸附能相比于Fe-N4/C结构没有增强效果,Fe―O和O―O键长也与未掺杂S的结构几乎相同。这是因为Fe-N4S4/C结构中S原子距离Fe-N4中心最远,对电子结构的影响很小,可认为该结构与不含S掺杂的Fe-N4/C结构对O2的吸附没有明显区别。
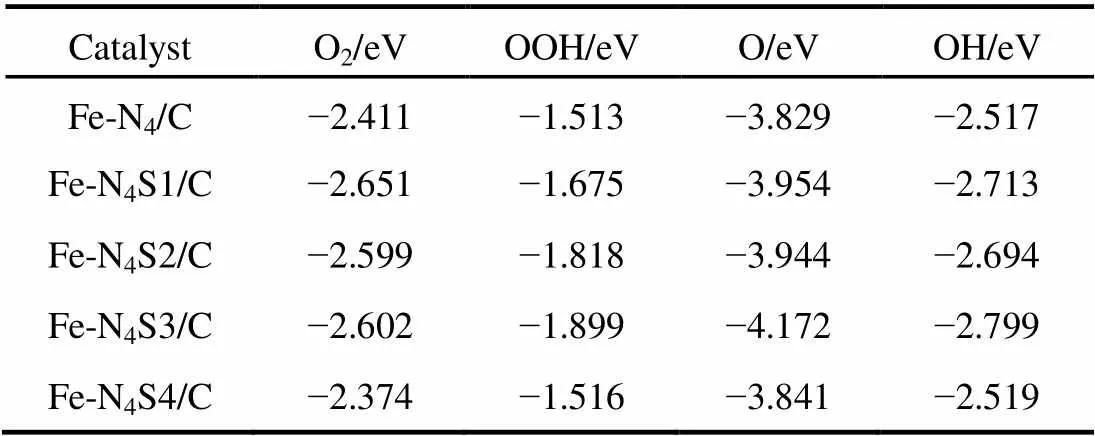
表3 O2,OOH,O,OH在不同的Fe-N4S/C结构上的吸附能

图8 Fe-N4S/C上吸附O2 (a), OOH (b), O (c), OH (d)的结构图
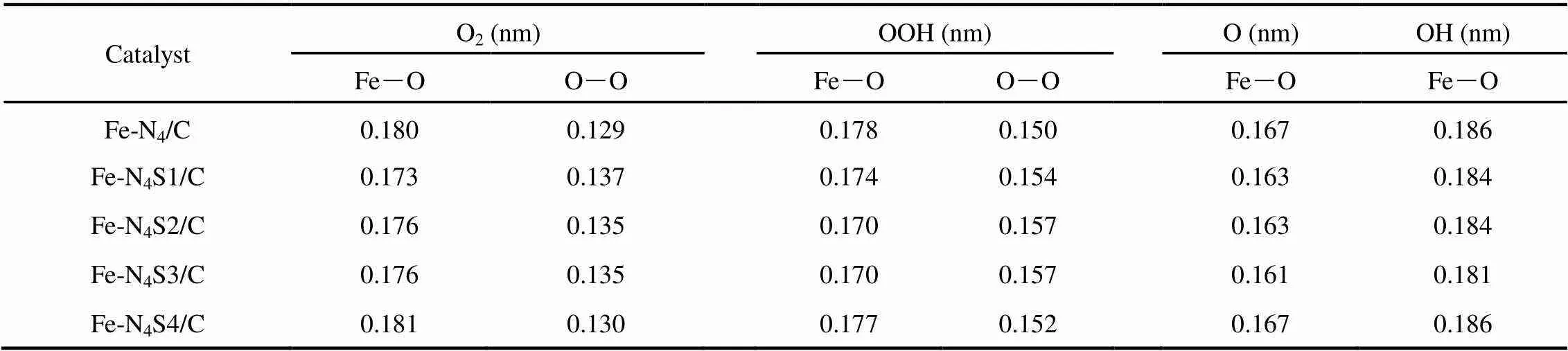
表4 O2, OOH, O, OH在不同的Fe-N4S/C结构上的原子间距
吸附的O2随后通过加氢变成OOH,OOH的吸附行为与O2相似,如图8(b)及表3和表4所示。在Fe-N4S1/C、Fe-N4S2/C和Fe-N4S3/C上OOH的吸附能比不含S掺杂的Fe-N4/C结构分别高出0.162 eV、0.305eV和0.386 eV,Fe―O键也相应变短。与O2相似,OOH在Fe-N4S4/C上的吸附能与在Fe-N4/C上的很接近。同时,OOH中O―O键的键长相比于孤立状态的O2也发生了明显的变化。OOH中吸附在Fe-N4/C结构上的O―O键长为0.150 nm,而吸附在Fe-N4S/C结构中的O―O键长分别为0.154 nm (Fe-N4S1/C),0.157 nm (Fe-N4S2/C),0.157 nm (Fe-N4S3/C),0.152 nm (Fe-N4S4/C),Fe-N4环外的S原子对于OOH的吸附影响仍然很小。这一结果表明,由于Fe-N4环内S原子的存在,增强了O2和OOH与Fe-N4结构形成Fe―O键的相互作用,较强的化学吸附导致O―O键键能显著降低,键长拉伸,为后续反应O―O键的断裂提供可能。O―O键被削弱得越多,越有利于ORR反应的进行。随后,OOH继续加氢后脱去H2O分子形成吸附O,吸附的O加氢形成OH,随后加氢形成第二个H2O分子。尽管O和OH在Fe-N4S1/C、Fe-N4S2/C、Fe-N4S3/C上的吸附也比在Fe-N4/C和Fe-N4S4/C上的强,但是由于其并不是决速步,因此对于ORR反应的顺利进行并没有太大影响。掺杂在Fe-N4环外的S原子由于与Fe原子中心的距离较远,对ORR催化活性没有明显的影响。
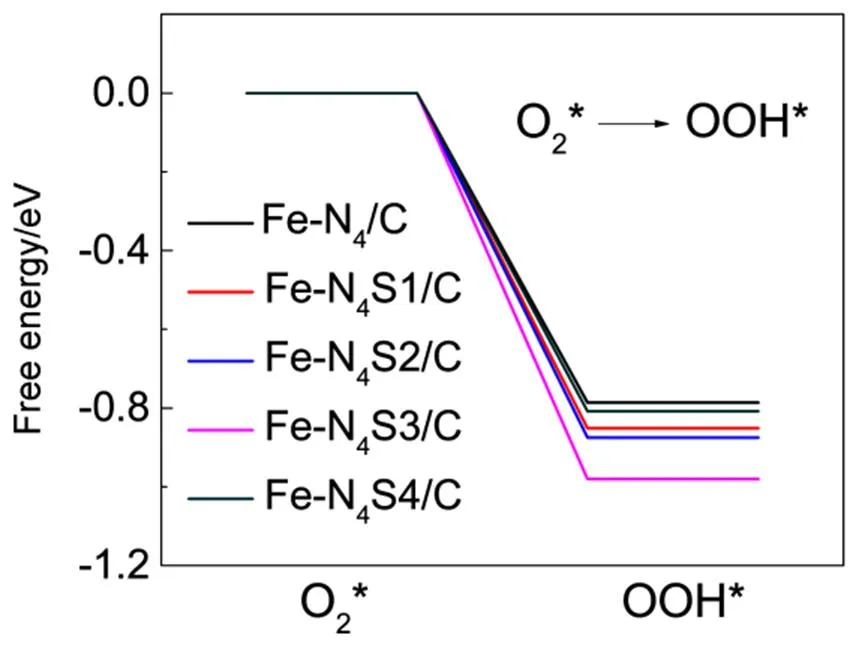
图9 Fe-N4/C和Fe-N4S/C上吸附的O2加氢的反应自由能曲线
O2的第一步加氢生成OOH(式5)通常被认为是ORR过程的速率决定步骤。进一步通过对比O2在Fe-N4/C及Fe-N4S/C结构上加氢过程的自由能能变来分析不同掺杂位置的S原子对ORR催化性能的影响。自由能能变越低,表明ORR过程越容易进行,催化活性越高。如图9所示,在Fe-N4S1/C、Fe-N4S2/C和Fe-N4S3/C中,O2加氢的自由能分别为−0.851 eV、−0.875 eV和−0.980 eV,相比Fe-N4/C结构(−0.786 eV)显著降低,表明在这三个位置上掺杂的S原子对于Fe原子中心催化ORR有较明显的促进作用。而在Fe-N4S4/C结构中,O2加氢的自由能仅稍低于Fe-N4/C结构,为−0.808 eV,对ORR的促进作用较小。这与通过吸附能计算得到的结果基本一致。
4 结论
本文通过改变前驱体中KSCN的添加量,在Fe/N/C催化剂中额外掺杂S元素,研究S掺杂对氧还原催化活性的促进作用,并通过催化剂的形貌结构、组成、表面特性等性质,结合密度泛函理论计算分析了S掺杂促进Fe/N/C催化剂的氧还原活性的原因。结果表明,适量的S元素可促使前驱体中过量的Fe形成FeS,防止形成单质Fe或含Fe纳米颗粒,从而提高催化剂的比表面积,促进传质,并实现较高的S掺杂含量,显著提高ORR活性。然而,过多的S则会形成过量的FeS,经酸处理后溶解,导致催化剂中Fe和S的掺杂含量同时降低,对活性不利。DFT计算结果表明,在Fe-N4结构内掺杂S原子可增强Fe原子中心与O2分子和中间体OOH的相互作用,促进形成Fe―O键,并削弱O―O键,导致O―O键的键能降低,更容易断裂,有利于ORR反应的进行。掺杂位置距Fe-N4结构较远的S原子则对活性没有明显影响。该研究结果对于理解非贵金属催化剂的活性中心,进一步理性设计高性能的氧还原催化剂有积极作用。
(1) Wang, Y.; Chen, K. S.; Mishler, J.; Cho, S. C.; Adroher, X. C.2011,, 981. doi: 10.1016/j.apenergy.2010.09.030
(2) Guo, S. J.; Zhang, S.; Sun, S. H.2013,, 8526. doi: 10.1002/anie.201207186
(3) Morozan, A.; Jousselme, B.; Palacin, S.2011,, 1238. doi: 10.1039/c0ee00601g
(4) Shao, M. H.; Chang, Q. W.; Dodelet, J. P.; Chenitz, R.2016,, 3594. doi: 10.1021/acs.chemrev.5b00462
(5) Lefèvre, M.; Proietti, E.; Jaouen, F.; Dodelet, J. P.2009,, 71. doi: 10.1126/science.1170051
(6) Proietti, E.; Jaouen, F.; Lefèvre, M.; Larouche, N.; Tian, J.; Herranz, J.; Dodelet, J. P.2011,, 416. doi: 10.1038/ncomms1427
(7) Tian, J.; Morozan, A.; Sougrati, M. T.; Lefèvre, M.; Chenitz, R.; Dodelet, J. P.; Jones, D.; Jaouen, F.2013,, 6867. doi: 10.1002/anie.201303025
(8) Tao, L.; Wang, Q.; Dou, S.; Ma, Z. L.; Huo, J.; Wang, S. Y.; Dai, L. M.2016, 2764. doi: 10.1039/c5cc09173j
(9) Choi, C. H.; Chung, M. W.; Park, S. H.; Woo, S. I.2013,, 1802. doi: 10.1039/c2cp44147k
(10) Wang, S. Y.; Zhang, L. P.; Xia, Z. H.; Roy, A.; Chang, D. W.; Baek, J. B.; Dai, L. M.2012,, 4209. doi: 10.1002/anie.201109257
(11) Xue, Y. Z.; Wu, B.; Bao, Q. L.; Liu, Y. Q.2014,, 2975. doi: 10.1002/smll.201400706
(12) Zheng, Y.; Jiao, Y.; Ge, L.; Jaroniec, M.; Qiao, S. Z.2013,, 3192. doi: 10.1002/anie.201209548
(13) Tylus, U.; Jia, Q. Y.; Strickland, K.; Ramaswamy, N.; Serov, A.; Atanassov, P.; Mukerjee, S.2014,, 8999. doi: 10.1021/jp500781v
(14) Wu, G.; More, K. L.; Johnston, C. M.; Zelenay, P.2011,, 443. doi: 10.1126/science.1200832
(15) Wang, Q.; Zhou, Z. Y.; Lai, Y. J.; You, Y.; Liu, J. G.; Wu, X. L.; Terefe, E.; Chen, C.; Song, L.; Rauf, M.; Tian, N.; Sun, S. G.2014,, 10882. doi: 10.1021/ja505777v
(16) Wang, Y. C.; Lai, Y. J.; Song, L.; Zhou, Z. Y.; Liu, J. G.; Wang, Q.; Yang, X. D.; Chen, C.; Shi, W.; Zheng, Y. P.; Rauf, M.; Sun, S. G.2015,, 9907. doi: 10.1002/anie.201503159
(17) Ferrandon, M.; Kropf, A. J.; Myers, D. J.; Artyushkova, K.; Kramm, U.; Bogdanoff, P.; Wu, G.; Johnston, C. M.; Zelenay, P.2012,, 16001. doi: 10.1021/jp302396g
(18) Liang, J.; Jiao, Y.; Jaroniec, M.; Qiao, S. Z.2012,, 11496. doi: 10.1002/anie.201206720
(19) Kattel, S.; Wang, G. F.2013,, 10790. doi: 10.1039/c3ta12142a
(20) Wu, G.; Santandreu, A.; Kellogg, W.; Gupta, S.; Ogoke, O.; Zhang, H. G.; Wang, H. L.; Dai, L. M.2016,, 83. doi: 10.1016/j.nanoen.2015.12.032
(21) Yang, X. D.; Zheng, Y. P.; Yang, J.; Shi, W.; Zhong, J. H.; Zhang, C. K.; Zhang, X.; Hong, Y. H.; Peng, X. X.; Zhou, Z. Y.; Sun, S. G.2017,, 139. doi: 10.1021/acscatal.6b02702
(22) Zitolo, A.; Goellner, V.; Armel, V.; Sougrati, M. T.; Mineva, T.; Stievano, L.; Fonda, E.; Jaouen, F.2015,, 937. doi: 10.1038/NMAT4367
(23) Chen, C.; Zhou, Z. Y.; Wang, Y. C.; Zhang, X.; Yang, X. D.; Zhang, X. S.; Sun, S. G.2017,, 673. doi: 10.1016/S1872-2067(17)62807-9
(24) CP2K (Open Source Molecular Dynamics). Available online: http://www.cp2k.org (accessed on 4 May 2017).
(25) Goedecker, S.; Teter, M.; Hutter, J.1996,, 1703. doi: 10.1103/PhysRevB.54.1703
(26) Hartwigsen, C.; Goedecker, S.; Hutter, J.1998,, 3641. doi: 10.1103/PhysRevB.58.3641
(27) Lippert, G.; Hutter, J.; Parrinello, M.1997,, 477. doi: 10.1080/002689797170220
(28) Deng, D. H.; Yu, L.; Chen, X. Q.; Wang, G. X.; Jin, L.; Pan, X. L.; Deng, J.; Sun, G. Q.; Bao, X. H.2013,, 371. doi: 10.1002/anie.201204958
(29) Xiao, M. L.; Zhu, J. B.; Feng, L. G.; Liu, C. P.; Xing, W.2015,, 2521. doi: 10.1002/adma.201500262
(30) Wohlgemuth, S. A.; White, R. J.; Willinger, M. G.; Titirici, M. M.; Antonietti, M.2012,, 1515. doi: 10.1039/c2gc35309a
(31) Wu, M.; Wang, J.; Wu, Z. X.; Xin, H. L.; Wang, D. L.2015,, 7727. doi: 10.1039/c4ta06323f
(32) Zhang, L. P.; Niu, J. B.; Li, M. T.; Xia, Z. H.2014,, 3545. doi: 10.1021/jp410501u
(33) Zhang, L. P.; Xia, Z. H.2011,, 11170. doi: 10.1021/jp201991j
(34) Wang, J.; Li, L.; Wei, Z. D.2016,, 321. [王 俊, 李 莉, 魏子栋. 物理化学学报, 2016,, 321.] doi: 10.3866/PKU.WHXB201512091
Experimental Boosting of the Oxygen Reduction Activity of an Fe/N/C Catalyst by Sulfur Doping and Density Functional Theory Calculations
CHEN Chi1,2ZHANG Xue2ZHOU Zhi-You2ZHANG Xin-Sheng1,*SUN Shi-Gang1,2,*
(1;2)
S doping in Fe/N/C non-precious metal catalysts is an effective approach to further improve their catalytic activity for the oxygen reduction reaction (ORR). However, the enhancement mechanism is not yet clear. Here, we synthesized an Fe/N/C catalyst using melamine-formaldehyde resin as the N and C precursors, CaCl2as the template, and FeCl3as the Fe precursor. The effects of S doping on the morphology, textural property, composition, and ORR catalytic activity were investigated by adding various amounts of KSCN as a precursor. Transmission electron microscopy (TEM) and N2adsorption- desorption isotherm results revealed that S prevented the growth of Fe-containing nanoparticles, and facilitated the formation of a porous structure, which increased both the catalyst surface area and mass transfer rate. X-ray photoelectron spectroscopy (XPS) results indicated that a suitable amount of S precursor led to a high doping level of S and provided the highest ORR activity. However, too much S in the precursor decreased the doping levels of both Fe and S, due to the formation of FeS, which could be completely removed by acid leaching. Density functional theory (DFT) calculations showed that the addition of S in an Fe-N4macrocycle could enhance the interaction strength of the Fe―O bond between the O2molecule or the intermediate OOHspecies and Fe in the Fe-N4structure, resulting in a significant decrease in the O―O bond energy, and may help in bond breaking in subsequent reactions, facilitating the ORR process.
Oxygen reduction reaction; Non-precious metal catalyst; Fe/N/C materials; S-doping; Density functional theory
March 30, 2017;
April 28, 2017;
May 8, 2017.
ZHANG Xin-Sheng, Email: xszhang@ecust.edu.cn; Tel: +86-21-64253469. SUN Shi-Gang, Email: sgsun@xmu.edu.cn; Tel: +86-592-2180181.
10.3866/PKU.WHXB201705088
O646
The project was supported by the National Natural Science Foundation of China (21373175 and 21621091).
国家自然科学基金(21373175和21621091)资助项目
猜你喜欢
杂志排行

物理化学学报的其它文章
- Development and Validation of a Reduced Chemical Kinetic Mechanism for HCCI Engine of Biodiesel Surrogate
- Cobalt@cobalt Carbide Supported on Nitrogen and Sulfur Co-Doped Carbon: an Efficient Non-Precious Metal Electrocatalyst for Oxygen Reduction Reaction
- 非离子表面活性剂Triton X-100溶液在不同生长期小麦叶片表面的润湿行为
- Efficient Synthesis of Sulfur and Nitrogen Co-Doped Porous Carbon by Microwave-Assisted Pyrolysis of Ionic Liquid
- First-Principles Study: the Structural Stability and Sulfur Anion Redox of Li1−xNiO2−ySy
- OL负载Cu催化剂及其催化氧化CO及乙酸乙酯性能