信号转导通路与肺纤维化关系的研究进展
2018-01-10宋玲莉李海陶晓根菅向东
宋玲莉 李海 陶晓根 菅向东
·综述·
信号转导通路与肺纤维化关系的研究进展
宋玲莉1,2李海1,3陶晓根1,4菅向东1
肺纤维化是多种原因引起的慢性肺疾病的共同结局,其病变特点是肺部炎症导致肺泡持续性损伤、反复破坏、修复、重建及细胞外基质(extra cellular matrix,ECM)的过度沉积,涉及到细胞、细胞因子、ECM 等因素间的相互作用,是多因素、多环节参与的缓慢的错综复杂的过程。信号转导通路在肺纤维化发生中起到的作用,成为近几年研究和探索的热点,细胞因子通过自分泌或旁分泌方式发挥其生物学作用,通过与其靶细胞表面受体相互作用将信号转导至细胞内,启动信号转导级联,调控基因的表达。笔者就参与肺纤维发生过程中主要信号通路的结构及功能和作用机制加以综述,以对肺纤维化的分子机制有全面的了解和认识。
一、mTOR信号转导通路
1.结构和功能
哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)是一种相对保守的丝氨酸/苏氨酸蛋白激酶,是磷脂酰肌醇激酶相关激酶(phosphatidy-linositol kinase-related kinase,PIKK)蛋白质家族的成员之一。mTOR可以对mRNA转录、代谢、自噬产生影响,从而调控细胞的生长。在酿酒酵母的雷帕霉素靶蛋白中包括两种多蛋白复合体:TORC1和TORC2。TORC1包含TOR1或TOR2,KOG1(YHR186c)和LST8。TORC2包含TOR2,AVO1,AVO2,AVO3和LST8。TORC1提升雷帕霉素灵敏度,调节TOR共享途径。TORC2降低雷帕霉素灵敏度,调节TOR2独立途径。因此,不同的TOR复合物确保了调节的特异性、多样性以及对雷帕霉素靶蛋白信号的选择性抑制。从酵母到人类,这两种蛋白复合体在进化中相对保守[1]。哺乳动物雷帕霉素靶蛋白分子量为289 000,也有两种多蛋白复合体,分别为mTORC1和mTORC2[2]。mTORC1由mTOR、raptor、mLST8以及负性调节因子PRAS40和DEPTOR组成,对雷帕霉素敏感[3]。mTORC2由mTOR、mLST8、rictor、mSin1、Hsp70、Protor和DEPTOR几个亚基组成[4-6]。雷帕霉素的长期干预可在一定程度上抑制mTORC2的合成,但整体来说,mTORC2较mTORC1表现出更多的雷帕霉素耐受性[7]。
mTORC1和mTORC2在信号转导中起到不同的作用。mTORC1可以调节细胞中蛋白质、脂类、细胞器的生物合成,同时可限制诸如自噬等异化作用。通过促进合成代谢过程,mTORC1可正向调节细胞的生长和增殖过程。对于mTORC1功能的了解大部分来源于细菌的大环内酯物雷帕霉素的使用。雷帕霉素进入细胞后,即与12 000的FK506结合蛋白质(FKBP12)结合并继续与mTOR的FKBP12-雷帕霉素结合区域(FRB)反应,从而抑制mTORC1的功能[8]。但FKBP12无法与mTORC1反应,或者不能起到强烈抑制mTORC1的作用[4,9]。以上信号通路显示,mTORC1对于雷帕霉素敏感,而mTORC2对于雷帕霉素不敏感。但另有研究显示长时间的慢性雷帕霉素治疗,在一定程度上可阻断mTORC2合成,从而对mTORC2活动起到抑制作用[7]。同时,Choo等[10]的研究显示,mTORC1对于雷帕霉素的抑制作用也存在一定抗性。
2.mTOR的信号转导途径
mTOR信号通路有多种上游调控途径,其中最主要包括IGF/PI3K/Akt/mTOR信号传导通路、TSC1-Tsc2/Rheb/mTOR信号传导通路、AMPK/TSC/mTOR信号通路等。在IGF/PI3K/Akt/mTOR信号传导通路中,细胞生长因子等激活上游反应元件磷脂酰肌醇3激酶后,产生第二信使—三磷脂酰肌醇(PIP3),PIP3随后与细胞内的AKT蛋白以及磷酸肌醇依赖性蛋白激酶(PDK1)结合,进一步磷酸化Akt蛋白的Ser308将Akt蛋白活化。而Akt可直接将mTORC1的SER2448位点磷酸化,激活mTORC1。TSC1-Tsc2/Rheb/mTOR信号传导通路中,其激活的初期路径与IGF/PI3K/Akt/mTOR信号传导通路类似,后续路径中,激活的Akt抑制结节性硬化症复合物1/2(tuberous sclerosis complex 1/2,TSC1/2),从而降低小G蛋白Rheb(Ras homologue enriched in brain)的抑制作用,使其从GDP结合状态转变为GTP结合状态。活化的Rheb(GTP结合状态)激活mTORC1。AMPK/TSC/mTOR信号通路中,当细胞中ATP/AMP比例降低时,AMP与AMP-相关蛋白激酶(AMPK)的亚基单位结合引起构象的变化,随后抑癌基因丝氨酸-苏氨酸激酶11(LKB1)和AMPKα亚基结合,磷酸化AMPK致其激活。激活后的AMPK可直接抑制mTORC1[11],亦可磷酸化TCS2。活化后的TCS2对Rheb产生抑制作用,使其从GTP结合状态转变为GDP结合状态,从而抑制了mTORC1活性[12]。mTOR信号通路下游调控主要包含蛋白质合成、自噬、脂类合成、线粒体的代谢与生物合成等。
mTORC1可正向调节蛋白质的合成。其方法包括磷酸化真核翻译起始因子4E(the eukaryotic initiation factor 4E,eIF4E)结合蛋白1(4E-BP1),以及p70核糖体S6激酶1(S6K1)。mTORC1对4E-BP1的磷酸化使其失去了与eIF4E结合的能力,从而使eIF4E处于游离状态,游离的eIF4E可与eIF-4A、eIF-4B、eIF-4G结合,形成了可与mRNAs 5’末端帽结构结合的eIF-4F,从而启动了帽依赖性翻译[13]。而mTORC1也可将PDK1磷酸化,进一步活化S6K1。S6K1的活化可激活核糖体蛋白S6,促进核糖体蛋白的生物合成;S6K1的活化也可激活SKAR、eIF4B,抑制PDCD4和eEF2K,从而促进mRNA生物合成、延伸、帽依赖翻译[14]。
3.与肺纤维化的关系
研究显示,AP24534能够通过AKT/mTOR/S6通路对肌成纤维细胞的活化进行调控,抑制TGF-β1诱导的肌成纤维细胞活化过程[15]。PQ可诱导肺纤维化,而雷帕霉素治疗可通过抑制mTOR信号通路,抑制TGF-β1和α-SMA的表达,从而有效抑制PQ诱导的肺纤维化,包括肺泡塌陷和胶原沉积等[16]。加减补肺汤可抑制小鼠肺组织中mTOR信号通路的过度表达,从而抑制博来霉素诱导的小鼠肺纤维化[17]。针对煤工尘肺患者的研究显示,持续接尘者的mTOR表达量要高于间断接尘者,脱尘1~10年的煤工尘肺患者肺泡巨噬细胞中mTOR和p-mTOR的表达量高于脱尘11~20年的煤工尘肺患者,巨噬细胞的自噬能力降低,该情况可能是影响肺纤维化进展的原因之一[18]。
雷帕霉素可提升肺纤维化小鼠的存活率,但其可被作为自噬抑制剂的氯喹所抑制。mTOR的过度激活是包括肺泡上皮细胞和成纤维细胞等多细胞诱导的肺纤维化的原因之一,肺纤维化的发病机制中包含肺泡上皮细胞mTOR的过度激活以及自噬能力的降低。抑制mTOR的活性以及诱导自噬能力可能是对IPF治疗的一种合理方法[19]。TGF-β1在体内至少部分通过激活mTOR通路来抑制成纤维细胞中的自噬作用,由TGF-β1导致的自噬能力降低可能是推动肺部特发性纤维化发生的机制之一。而雷帕霉素的使用可降低α-平滑肌肌动蛋白和纤维联接蛋白的表达,产生抗纤维化作用[20]。
作为extra type III domain(EDA)剪切和成纤维细胞激活的潜在诱导因子,TGF-β,抑制了PTEN在间充质细胞中的活性和表达,增强了PI3K/Akt信号。pten-/-细胞中,mTOR信号的抑制也阻断了FN的产生以及EDA的剪接,导致由Akt介导的EDA剪接调节蛋白SF2/ASF磷酸化受到抑制。pten-/-细胞中的纤维连接蛋白停止表达导致了细胞繁殖和迁移能力的降低。PI3K/Akt/mTOR有利于FN转录和选择性剪接,从而调节细胞的行为[21]。
二、Notch信号通路
1.结构与功能
Dexter在1914年发现部分果蝇翅缘出现缺口现象。1917年,Morgan[22]在果蝇体内发现一种基因,该基因功能部分缺失可导致果蝇翅缘出现缺口,因此将其命名为Notch基因。而Artavanis-Tsakonas等[23]首次对该基因进行了克隆。研究显示,Notch基因家族具有高度的保守性,在细胞分化和发育中起到关键作用[24]。Notch信号通路由Notch配体、Notch受体、CSL-DNA结合蛋白等构成[25]。
果蝇Notch配体包括Delta和Serrate,线虫Notch配体是Lag-2,取Delta、Serrate和Lag-2的首位字母,称Notch配体为DSL蛋白。目前在哺乳动物共发现了5个同源配体,同源配体分为两类:与Delta同源性高的配体被称作Delta或Delta-like(Dll),分别为Dll1、Dll3、Dll4;与Serrate同源性高的配体被称作Jagged,分别为Jag1和Jag2。
Notch的配体是Ⅰ型跨膜蛋白,胞外区具有数目不等的表皮生长因子(epidermal growth factor,EGFR)样重复序列和一个N末端富含半胱氨酸的DSL区域,DSL区域在配体家族中高度保守,与Notch受体结合后使之激活[26]。Jagged1、2的胞外序列同样存在以上两个区域,同时Jagged配体还包括一个半胱氨酸的基序富集区域,而Delta配体胞外序列无此区域。Notch配体的胞内域较短,约有70个左右氨基酸残基,功能机制有待进一步研究确认。
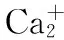
CSL是CBF1、Su(H)、Lag-1三者首字母的缩写,CBF1、Su(H)、Lag-1分别是CSL在哺乳动物、果蝇、线虫的不同名称,他们是Notch信号在核内活化的转录因子,在哺乳动物CBF1蛋白也被称为RBP-J。CSL是Notch信号通路的初级效应分子,是转录抑制因子,可与Notch受体的胞内区的RAM和Ank序列相互结合,形成复合物,使CSL从转录抑制剂转变为转录激活剂,进而激活下游靶基因的表达。
Notch信号途径的靶基因有多种,在哺乳动物中是HES家族。HES基因编码bHLH类转录因子,其碱性结构域可与DNA相结合。哺乳动物的HES家族中的HES-1和HES-5在Notch信号通路中发挥作用[28]。
经典Notch信号通路分子除了包括Notch配体、Notch受体和CSL-DNA结合蛋白外,还包括其他一些修饰分子,如糖基转移酶Fringe、膜相关蛋白Numb、mastermind、TLE/Groucho蛋白,它们对Notch信号转导起协同激活或者抑制作用。
2.信号转导途径
Notch的激活方式:Notch蛋白从初始合成到具有生物学功能的过程需要经历3次切割。Notch首先在高尔基体内完成合成后,在SI位点处被furin样转化酶切割,切割后形成的NECD和NTMIC,两者通过非共价键结合在一起,形成NECD-NTMIC二聚体,随后转运至细胞膜上。Notch受体与配体结合后,导致受体细胞外S2切割位点的暴露,S2位点位于跨膜区域前的大约12个氨基酸处,且深埋在负调节区域中。S2位点的水解是Notch激活过程中非常重要的一步,主要酶切方式是在ADAM金属蛋白酶家族的TACE或Kuz酶介导的水解作用下,与S2位点进行酶切,notch受体释放部分胞外片段,将剩余部分连接在细胞膜上,这种结构称为Notch-intro[29]。最后,早老素在与Nicast rin、APH-1和PEN-2三种蛋白形成一个多蛋白复合体后,激活γ-促分泌酶,对S3位点进行酶切,使North释放其胞内区(Notch intracellular domain,NICD)[30]。NICD转移至细胞核内,其RAM区域首先与DNA结合蛋白CSL(CBF1/RBPjκ/Su(H)/Lag-1)结合。NICD的ANK区域随后与CSL结合激活MAML,形成激活复合体,作用于靶基因HES和HRT等,该类基因多为碱性螺旋环螺旋(basic helix-loop-helix,bHLH)结构的转录抑制因子,调节下游基因的转录和表达[31]。除了经典的Notch-CSL途径外,Notch信号还可通过不依赖CSL的途径进行[32]。研究表明一种泛蛋白化跨核膜的蛋白质DTX(Deltex)可以激活Notch,DTX由胞内部分和核内部分构成。作为DTX的同系物DTX1,其核内部分可以通过与转录辅助激活因子P300的结合,抑制促进型bHLH基因Mash-1的活化,将神经干细胞维持在未分化状态[33]。
3.与肺纤维化的关系
Notch信号通路调节的肺纤维化机制是一个复杂的病理生理过程,是信号通路与各种细胞因子形成了信号网络,共同调控肺纤维化的进程。而以细胞外基质ECM的过度沉积为核心的,包括成纤维细胞灶的形成、肺泡上皮损伤等,是该进程的重要病理特征。
研究表明,Notch信号通过炎症细胞因子调节肺纤维化的病理生理过程。Notch信号通路通过炎症细胞因子调控肺纤维化肺纤维化的形成与多种炎症细胞因子有关,炎症与抗炎平衡的失调可以加速肺纤维化的进展。转化生长因子-β(transforming growth factor-β,TGF-β)属于调节细胞生长和分化的TGF-β超家族,是一类分布广泛的生长因子家族,其主要作用包括参与调节细胞增殖和分化,胚胎发育调节,以及促进细胞外基质ECM形成和抑制免疫反应等。同时其属于炎症细胞因子,在正常成纤维细胞发生表型转化时,可改变成纤维细胞贴壁生长的特性。Notch信号上调对肺泡上皮细胞中胶原和α平滑肌肌动蛋白(α-SMA)表达具有促进作用,导致肌纤维母细胞的分化进而影响肺纤维化的形成和发展,推测TGF-β和Notch信号相互关联作用调控肺纤维化进程[34-35]。而γ-分泌酶抑制剂(DAPT)的使用则可以下调Notch信号,并在一定程度上减轻肺组织纤维化[36]。同样在使用DAPT后,可以使在肌成纤维细胞(myofibroblast,MB)中较正常上皮细胞中明显升高的TGF-β mRNA及ECM降至相同水平[34]。推测Notch1可与TGF-β共同作用,对EMT起诱导作用,增加ECM的分泌量,NICD/RBP-Jk复合体在该信号通路中发挥重要作用;膜结合的TGF-β活化Notch1-HES1轴,释放的NICD进入细胞核,与DNA结合,激活下游调控基因的转录,增加了ECM的分泌,从而促进肺纤维化,而DAPT可Notch阻断剂阻止该反应的发生[37]。TNF-a是由多种细胞分泌的一种跨膜蛋白,其介导的信号传导主要通过Ⅰ型跨膜的糖蛋白受体(TNFR1)和Ⅱ型跨膜的糖蛋白受体(TNFR2)进行。Notch可以下调基质金属蛋白酶家族成员ADAM-17的活性,减少受体TNFR1的降解,并上调TRAF-2的活性,进一步促进TRAF-2与TNFR1特定部位的结合,诱导肺部炎症的加重,促进肺纤维化[38]。
发现于炎症区域1(Found in inflammatory zone 1,FIZZ1)具有生长因子和炎症因子的特性,具有促进细胞增殖、抗凋亡和趋化等作用。PI3K/Akt通路参与了FIZZ1介导的肺血管平滑肌细胞的增殖和迁移,PI3K的抑制剂可部分抑制FIZZ1引起的肺血管平滑肌细胞的增殖[39],而Notch通路也能介导FIZZ1的表达[39]。因此,对PI3K/Akt通路和Notch通路的共同抑制提升肺纤维化疾病的治疗效果。Notch信号可激活巨噬细胞的分化,导致单核巨噬细胞产生过量一氧化氮(NO),造成NO在肺内的过度充盈,可造成肺间质纤维化,应用DAPT及外源性诱导配体Jagged1-Fc对Notch信号通路进行抑制剂,进一步抑制巨噬细胞的分化增殖,从而减少NO的生成,抑制肺纤维化的发生[40]。同时肺部损伤时,伴随作为端粒酶催化亚单位的端粒酶反转录酶(telomerase reverse transcriptase,TERT)基因表达,激活端粒酶活性。而TERT在肺内的表达与Notch1呈正相关[41],Notch表达下调可以降低TERT表达,抑制端粒酶活性,从而减缓肺纤维化进程。
三、Wnt/β-catenin信号通路
1.结构与功能
Nusse等[42]于1982年从鼠类乳腺癌病毒(MMTV)诱导的小鼠乳腺癌中克隆出的一种原癌基因,时称int21基因。后来的研究发现[43],int基因与Sarma报道的果蝇无翅基因wingless(wg)为同源基因,因而将两者合称为Wnt。Wnt基因编码的Wnt蛋白家族属于分泌型糖蛋白,含一段信号肽以及23或24个位置保守的半胱氨酸残基,通过旁分泌或自分泌方式作用与位于细胞膜上的特异性受体相结合,激活胞内的各级信号转导分子,从而调节靶基因的表达。基于诱导小鼠乳腺上皮细胞系C57MG转化能力的差异,Wnt蛋白配体可分为两类,分别为:Wnt1类和Wnt5a类[44]。Wnt1类蛋白在胚胎形成早期可以诱导体轴及中枢神经系统的形成,包含Wnt1、Wnt3、Wnt3a、Wnt7a、Wnt8a和Wnt8b蛋白;而Wnt5a类包含Wnt2、Wnt4、Wnt5a、Wnt5b、Wnt6、Wnt7b和Wnt11蛋白。Wnt的受体分为三类:卷曲蛋白(Frizzled,Fz)、LRP5/6和Ror、Ryk家族[45-46]。卷曲蛋白是一种7次跨膜蛋白,结构类似于G蛋白偶联型受体,目前已知有10个家族成员Frz1~10。Frizzled蛋白共有4个重要的结构区,包括位于胞外细胞膜表面的N端、高度可变的亲水区、信号肽序列、以及120个氨基酸区段。其中半胱氨酸富集区(cysteinerichdomain,CRD)高度保守,胞外Wnt信号与卷曲蛋白CRD区结合并改变CRD区构型,从而诱导蓬乱蛋白(dishevelled,Dsd/Dvl)活化。Wnt的第二类受体LRP5/6,属于低密度脂蛋白受体相关蛋白(low density lipoprotein receptor related proteins,LRP)。Wnt的第3类受体Ror、Ryk家族,属酪氨酸激酶受体[47]。
2.信号路径
Wnt信号通路至少可分为三种类型:Wnt经典途径(Wnt/β-catenin pathway)、Wnt-Ca2+♂信号通路和平面细胞极性途径(planar cell polarity pathway,PCP)[48]。其中Wnt/β-catenin途径是研究最多也是最为深入的一条途径。PCP途径,即Wnt/PCP途径,在脊椎动物中又称Wnt/JUN激酶途径,该途径的主要作用可能与胎发育阶段调控细胞骨架的重排有关,并参与原肠胚的形成。Wnt-Ca2+♂途径[47],由Wnt-5α或Wnt-11激活卷曲蛋白受体后,由三聚体G蛋白介导,通过钙调蛋白依赖的激酶(CaMKⅡ)、蛋白激酶C(PKC)起作用,诱导细胞内Ca2+♂浓度增加,活化T细胞核因子,使核内NFAT增加而激活靶基因[45]。
经典Wnt信号通路中,(-catenin是最为关键和核心的信号转导因子[49],经典Wnt通路未被激活时,细胞质内大部分的β-Catenin与细胞膜上Cadherin蛋白结合而附着于细胞骨架蛋白肌动蛋白上,参与细胞的黏附作用;而细胞质内存在少部分的β-Catenin。此时,细胞质内存在的一种轴抑制因子(Axin)/GSK-3/APC复合物,该复合物以Axin为支架,具有多个与其它蛋白作用的位点,该类位点与APC、糖原合成酶激酶3β(glycogen synthase kinase 3β,GSK3β)及酪氨酸激酶1α(tyrosine kinase 1α,CK1α)结合,形成了Axin/GSK-3/APC复合物。其中,CK1α能将β-Catenin的Ser45位点磷酸化,随后GSK3β将β-Catenin的Thr41、Ser37、Ser33位点磷酸化。GSK3β是一种丝氨酸/苏氨酸蛋白激酶,能将磷酸基团加到β-Catenin N端的丝氨酸/苏氨酸残基上。APC蛋白既可与Axin反应,也可与β-Catenin反应。此时由于无Wnt/Wg配体存在,胞质中的Axin复合体处于稳定状态,复合体磷酸化β-Catenin,磷酸化的β-Catenin被E3泛素连接酶β-转导素重复序列包含蛋白(β-transducin repeat containing protein,β-Trcp)识别,泛素化后被蛋白酶体降解,使胞质中的β-Catenin 维持在较低的浓度,β-Catenin不能激活细胞核中的靶基因,靶基因转录处于沉默状态[50]。当细胞外存在Wnt蛋白时,细胞膜受体LRP5/6和Frizzled与Wnt蛋白结合,形成受体蛋白复合体[51]。受体蛋白复合体的胞内端将通过募集Axin和蓬乱蛋白1(scaffolding protein dishevelled 1,Dvl)等,Dvl的激活使得降解β-catenin的Axin/GSK-3/APC复合体解体,使GSK3β失去发挥其功能的结构支持,阻碍了GSK3β的磷酸化活性;也造成GSK3β磷酸酶活性直接失活,使得细胞内β-catenin蛋白得以积累并不断升高[52]。细胞质中不断累积的β-catenin逐渐转移至细胞核,与细胞核中转录因子T细胞因子/淋巴增强子因子(T-cell factor/lymphoid enhancer factor,Tcf/Lef)家族蛋白结合[46,53],(-catenin结合并激活Tcf/Lef后,Wnt的靶基因并不能立即被激活[54]。当激活型转录因子结合到DNA上后,转录因子需要同时招募一系列转录辅助因子,开启下游基因的转录。(-catenin蛋白可分为三个功能区域:C端、12个Armadillo重复区段(R1~R12)组成的中段和N端区域。C端区域为转录激活区域,可以结合一系列通用转录辅因子,促进转录的起始和延伸;重复区段的R3~R10区域可以介导(-catenin与TCF结合[55]。N端区域可以与辅因子Bcl9结合[56]。经典Wnt信号通路中,Dvl蛋白和Wnt蛋白等是Wnt信号通路的正向调节因子,而Axin、APC、GSK-3等是该信号通路的负向调节因子。正向或者负向调节交互作用而引起细胞内β-Catenin含量、分布以及功能的变化,进而激活或抑制下游靶基因表达的变化。
3.与肺纤维化的关系
Wnt/β-catenin信号通路在细胞的增殖和变异中发挥了重要的作用,并且Wnt/β-catenin信号通路的异常活化与特发性肺间质纤维化(idiopathic pulmonary fibrosis,IPF)的形成有密切关系[57]。IPF是一种特殊类型原因不明的、发生于成人的、慢性、进行性、纤维化性间质性肺炎[58],其特征是纤维母细胞的异常增生及肺组织损失[59]。(肌)成纤维细胞活性的增加,ECM聚集,肺泡II型上皮(ATII)细胞功能异常均可作为IPF的发病机制。无论在小鼠肺纤维化模型中还是人发生IPF时,ATII细胞中的Wnt/β-catenin信号通路都会被激活,导致Wnt靶基因编码的WISP1表达,可导致ATII细胞增殖和上皮间质转化(epithelial-mesenchymal transition,EMT)。而使用WISP1抗体进行中和治疗后,可明显减缓肺的纤维化,包含降低胶原沉积,提升肺的功能以及存活率[60]。
Wnt信号无节制的激活是一种肺损伤或者修复程序的生理反应。经典Wnt信号通路中,某些组分的损伤性诱导、自发性突变,或者调控分子的信号调节,均会导致细胞增殖调节异常。Chilosi等[61]的研究显示,其余肺增生型疾病,甚至是IPF中,细胞核中β-catenin的上调是一个应答程序而非主要动因。伴随着β-catenin浓度增加而出现的二型上皮细胞和间质成纤维细胞的无调控增殖,提示在肺疾病中,相关信号通路(例如IPF,Wnt信号通路)一直处于激活状态。而Wnt信号通路抑制剂是一种控制这种增生的有效方法。此时,气道上皮细胞相邻的肌成纤维细胞的细胞核中β-catenin浓度上升。多项证据显示,无论在体外还是体内,肌成纤维细胞均可能通过降解细胞间质的方法诱导与其相邻的气道上皮细胞凋亡[62-64]。Chilosi等[61]对20例IPF/UIP肺样本的研究显示,18例样本中细胞核β-catenin出现积聚现象,Wnt信号通路调控的两个下游靶基因:cyclin-D1和matrilysin均存在异常表达。同时在IPF/UIP肺样本中β-catenin浓度增加通常会伴随细支气管病变,而该情况在其他类型肺疾病或者肺纤维化疾病中并未出现。研究发现在IPF中,Wnt基因,包括Wnt2、Wnt5a、Fzd7、Fdz10,会伴随着sFRP1和sFRP2出现过表达现象,而在正常肺组织以及其他间质性肺病中,该情况未出现[65]。更重要的是,在增生支气管损伤(基底细胞增生、鳞状上皮化生、支气管扩张和形成蜂窝状)中确认了细胞核β-catenin的免疫反应性以及细胞周期蛋白D和机制溶解因子浓度异常[61]。在大部分IPF样本的成纤维细胞中,细胞核中的β-catenin出现了积聚。该情况通常与支气管病变相关联,并且在肺纤维化试验中显示了与之类似的结果[61,66]。随后研究显示,Wnt1、7b和10b,Fzd2和3,β-catenin以及LEF1在IPF患者中显著表达;而通过使用qRT-PCR对原发性肺泡Ⅱ型上皮细胞的研究显示,Wnt1,Wnt3a,β-catenin和GSK-3β主要集中与肺泡细胞和支气管上皮细胞中。在体外功能研究中发现,Wnt3a诱导了肺上皮细胞增生、(肌)成纤维细胞活化以及胶原增生[67]。亦有报道证明,在IPF患者的增生性肺泡上皮细胞中出现了DKK1-4的拮抗作用,而DKK1主要位于基底支气管上皮细胞中[68]。在系统性硬化肺纤维化疾病中,细胞核中的β-catenin出现了增加,并加速了肺纤维化的转移和扩散;呼吸设备激活的Wnt/β-catenin信号通路与呼吸机诱导的健康肺部的肺纤维化相关[69]。
四、Hedgehog信号通路
1.结构与功能
Hedgehog(Hh)基因是1980年由Nüsslein-Volhard和Eric F.Wieschaus[70]在研究黑腹果蝇时作为一个“体节极性”基因发现的,因该基因的突变可导致果蝇胚胎呈多刺球状,与刺猬类似,因此命名为“Hedgehog”。在Mohler、Forbes等[71-72]的研究结果显示有相关基因共同参与了Hh信号的转导过程,逐步建立了Hh信号通路模型。
Hh信号通路由Hedgehog(Hh)、2个跨膜受体即Patched(Ptch)和Smoothened(Smo)、下游转录因子Gli家族等组成。Hedgehog基因编码一种分泌性信号蛋白,可以介导细胞间相互作用[73]。果蝇只有一个Hh基因;脊椎动物中的鸟类和哺乳动物中发现了3个Hedgehog同源基因,可编码三种Hh蛋白,即Shh、Ihh和Dhh;脊椎动物中的鱼类有6个Hedgehog同源基因,除了Shh、Ihh、Dhh外,还包含Twhh,Ehh和Qhh[74]。Shh的表达贯穿于脊椎动物的整个神经系统的发育过程,也存在于很多皮组织中;Ihh的主要功能主要集中于骨骼的发育;而Dhh的表达则主要限于外周神经系统和生殖器官[73]。Hh信号在发育过程中的缺失或减少,能导致多种缺陷和畸形的发生[75]。Hh蛋白由2个部分组成:羧基端(Hh-C)和氨基端(Hh-N),其中Hh-C具有自身蛋白水解功能及胆固醇转移功能,而Hh-N具有Hh蛋白的信号转导功能。Ptch蛋白是含12次跨膜结构域的膜蛋白,能与Hh蛋白结合,对Hedgehog信号通路起到负向调控作用。在人类中有两个Ptch同源基因,分别为Ptch1和Ptch2,其分别编码Ptch1和Ptch2蛋白。Smo蛋白定位于细胞膜,是一个7次跨膜蛋白,归属于G蛋白耦联受体FZ/SMO超家族成员,Smo有一个极长的C-末端胞内域,在Hh信号转导过程中发挥了极其重要的作用。在Hh信号转导中,Smo功能受Ptch影响,在信号转导中起到桥梁作用,其磷酸化水平影响细胞内相关分子的磷酸化反应,进而改变核转录因子的状态。Hh信号通路的转录因子Ci在果蝇中,仅有Ci一种核转录因子发挥了转录激活和抑制作用;而在哺乳动物对应于Ci转录因子的是Gli转录因子,哺乳动物的Gli具有锌指结构,分子量约为155KD。其中人类Gli基因家族包含3种同源基因:Gli1、Gli2和Gli3,Gli1、Gli2和Gli3蛋白,其相互之间存在协同与拮抗作用。Gli1始终起到转录激活的作用,Gli2主要生成转录激活子(GliA),而Gli3主要生成转录抑制子(GliR)。在Hh缺乏时,Gli3的C-末端会在GSK3β、PKA以及CK1的作用下发生磷酸化,磷酸化的Gli3会募集具有F-box亚基的SCF家族泛素连接酶β-TrCP,从而部分降解Gli3,生成Gli3R。该部分降解作用可能与Gli3的C-末端的PDD结构域有关[76]。Gli2中含有类似的PDD结构域,但其功能可能低于Gli3 PDD结构域,进而可能导致了GliR主要来源于Gli3,而非Gli2。当Hh存在时,阻断了Gli的部分水解作用,导致Gli2和Gli3进一步修饰形成转录激活子GliA,进入细胞核激活靶基因的表达。当Hh信号通路激活时,Gli1会大量表达,因此Gli1是经典Hh信号通路激活程度的一个重要标志物。
2.信号通路
Hh蛋白家族属于分泌性蛋白,约为45 000,由两个结构域组成,分别为羧基端和氨基端。当Hh蛋白转运进入内质网后,在Hh蛋白羧基端的胆固醇依赖性的自剪切作用下,生成了两个单独的片段,即19kDa的经胆固醇修饰的Hh-N和25kDa的Hh-C。该剪切过程需要经过两步方可完成。首先,蛋白的198位游离巯基半胱氨酸作为亲核试剂,进攻其之前的甘氨酸残基的羰基碳,形成硫酯中间体[77-80]。随后硫酯中间体受到胆固醇3β-羟基的亲核攻击,形成了替代C末端片段的胆固醇修饰的N末端片段。另外,363位的保守半胱氨酸在该剪切过程中亦起到重要作用,他与198位半胱氨酸形成二硫键,促进蛋白的折叠,并提供了剪切所需要的巯基,而其中任何一个半胱氨酸残基的突变均会阻止Hh自剪切[81]。随后,在Hh酰基转移酶Ski的催化作用下,Hh-N的N末端发生棕榈酰化[82]。因而,Hh-N具有两个共价结合的脂类基团:C-末端的胆固醇和N-末端的棕榈酸酯。最终,Hh-N获得了Hh所有的信号通路功能。Hh-N具有远距离传递达到其作用位点的功能,在哺乳动物肢体中该距离可达到300μm,而在果蝇翅盘中该距离也可达到50μm[83]。Hh-N的分泌和远距离信号传递需要在Disp活性作用下进行,Disp是属于RND家族的一种12次跨膜蛋白[84-85]。
Hh-C的胆固醇修饰对Hh多聚复合体的形成起到重要作用,该作用的过程尚不明确,其中一种可能性是将Hh锚定在细胞膜上,并将Hh聚集在脂质筏等特定微域,增加Hh单体间的静电作用[86]。胆固醇介导的聚集作用也可能促进Hh与肝素硫酸蛋白聚糖(HSPGs)等膜结合分子的反应[87-88],同时胆固醇也对Hh配体的浓度进行调节[89],但其调节机制尚不明确。未经胆固醇修饰的Hh配体仍具有一定的信号功能,但未经棕榈酰化的配体则几乎完全丧失信号功能[90-91],显示棕榈酰化是Hh信号通路所必需的。
Ptc与细菌转运蛋白RND家族具有同源性[92],是Hh配体在细胞膜上的主要受体。一些细胞膜上跨膜蛋白可以作为Hh的辅助受体,促进Hh与Ptc的结合。该类蛋白包含果蝇的Ihog和Boi蛋白以及脊椎动物中与之对应的Cdo和Boc蛋白,另外还包含脊椎动物特有的Gas1蛋白[93]。脊椎动物还存在一种可与Hh进行结合的蛋白Hip,该蛋白不具有信号传递功能,但其具有与Ptc类似的氨基酸序列,因此可与Ptc竞争性地结合Hh配体,对Hh信号转导起到负向调节作用。同时,通过抑制Smo活性,Ptc也可对Hh信号通路进行负向调节,但其抑制机制尚不明确[94]。在Hh配体处于缺乏的状态时,Ptc聚集于原纤毛,维持Smo的非激活状态,阻止Smo进入纤毛[95]。Hh与Ptc结合后使其失去活性,Smo以非激活状态进入原纤毛内,在磷酸化修饰后,形成激活状态进而激活下游信号通路[96]。而一些小分子,包含多巴胺和蒜藜芦碱等,均能影响Smo活性,其中多巴胺可以直接与Smo结合并阻止Smo在信号通路的激活作用[97]。Smo蛋白C-末端的磷酸化激活是由PKA和CK1α完成的,而GRK2可促进Smo向细胞膜聚集。虽然脊椎动物的C-末端与果蝇的相比,缺少PKA磷酸化位点,但其Hh信号仍可引起其磷酸化[98]。CK1α、GRK2在Smo磷酸化后会募集β-抑制蛋白和驱动蛋白2家族中的Kif3a,促进Smo进入纤毛[99],将Hh信号传递至核转录因子Gli上。在果蝇的Hh信号通路中,依赖于Hh配体的浓度水平,Ci同时表现出具有抑制和激活下游基因转录的功能。在Hh信号通路处于非活化状态时,蛋白水解酶可将Ci-FL水解,并生成N-末端转录抑制因子(Ci-R)进入细胞核,并抑制下游基因的转录功能。当信号通路处于活化状态时,Ci-FL的剪切作用受到抑制,经加工后生成转录激活因子Ci-A,进入核内,激活下游基因的转录。而与果蝇Ci因子对对应的脊椎动物的Gli所包含的三种因子分别为:Gli1、Gli2、Gli3。Gli2、Gli3具有转录激活和转录抑制的双重作用,而Gli1只起到激活作用。处于激活状态的Gli将促进包括分化、增殖等基因的转录表达,同时也对信号通路中的Ptc和Hip起到负向调节的作用,下调信号通路的活性。
3.与肺纤维化的关系
慢性呼吸道炎症可导致肺纤维化的发生。组织的重构和纤维化可以严重影响肺部功能。在组织重构过程中,包含上皮细胞、内皮细胞、成纤维细胞等的多种细胞发生了相互作用[100],当重构过程未能对组织完成修复,组织纤维化就会随着疤痕组织的形成而发生[101]。Shh和TGF-β等蛋白可以介导呼吸道的上皮细胞、内皮细胞、周皮细胞等结构细胞通过分子重排机制促进肺纤维化的发生[100,102-103]。Shh的旁分泌信号对胚胎时期肺的分支形态发生具有极其重要的作用[104]。在肺的分支形态发生过程中,内胚层制造了Shh,进而促进间充质细胞增生和分化。观察显示Shh的过度表达会导致肺间质的异常增加[105]。该类实验结果在一定程度上提高了Hh信号通路参与肺纤维化的可信度。在博莱霉素诱导的成人肺损伤模型中,肺泡隔膜中的大量细胞显示Gli1阳性,而纤维化损伤中Gli1阳性间充质细胞数量增加,并且腺病毒介导的Shh过表达增加了ECM的产生[106]。另有研究证实,在人肺部发生特发性肺纤维化、小鼠肺炎以及异硫氰酸荧光素诱导的纤维化时,Shh在肺上皮细胞中均出现过表达现象[100]。使用原位杂交的方法研究显示,肺囊肿上皮细胞中的Shh高度表达[107]。Bolanos等[108]的研究显示,IPF时的Hh信号通路出现激活现象,同时大量的体外研究数据显示Shh可以提高纤维母细胞的转移、扩散和存活,并导致ECM的产生。而Hh信号通路可增加ECM的产生并引起纤维母细胞向成肌纤维细胞转化也证实了该情况[109]。同时,研究人员在静息的外周血T淋巴细胞、浸润的单核细胞以及肺泡巨噬细胞中法相了Shh的受体Ptch[100]。在患有间质性肺病的患者中,这种重构的发生是连续的,并导致肺纤维化,同时伴随着包含T淋巴细胞和B淋巴细胞在内的明显的单核细胞浸润过程[110]。FOXF1是Shh在肺中对Gli2/3进行激活的下游作用位点[111-112]。但肺中的Shh和FOXF1在普通的间质性肺炎和非特异性的间质肺炎中的表达水平存在差异。据推测间质性肺炎的致病路径可能包含了在Hh信号通路中激活FOXF1的缺陷[107]。这些发现均证明Hh信号通路和其他各种因素共同作用导致了肺纤维化的发生。
五、TGF-β/Smad/ERK信号
1.结构与功能
转化生长因子-β(transforming growth factor-β,TGF-β)是生长因子超家族中具有免疫负调控功能的细胞因子家族,由Roberts等[113]从小鼠的肉瘤细胞中分离获得,是由二硫键相连形成的一种同源二聚体多肽分子,分子量25kDa,由于它能使大鼠肾脏细胞在琼脂上生长而得名[114],其对细胞生长、分化、细胞外间质增生、细胞凋亡、以及胚胎发育等均起到极其重要的作用[115-116]。在哺乳动物中至少发现了3种亚型:TGF-β1、TGF-β2、TGF-β3,其中对TGF-β1的研究最为透彻,它在人的血小板和哺乳动物的骨骼中含量最高。
新合成的TGF-β以非共价键与潜活性相关蛋白(LAP)结合形成没有活性的小复合物(SLC),该复合物可进一步与潜活性TGF-β结合蛋白(LTBP)以二硫键的形式结合,形成分子量约为225kDa的大复合物(LLC)。TGF-β以无活性的大复合物形式分泌到细胞外,与细胞上的受体结合从而发生生物学作用。TGF-β1的受体有3种,分别为TβRⅠ、TβRⅡ和TβRⅢ,它们都是单个跨膜α螺旋受体[117]。TβRⅠ主要包括TβR-I、ACTR-1、ActR-IB、XTr-I、ALR7、ATR-1、BMPR-1A[118],其结构由4部分组成:信号肽、亲水胞外区、跨膜区和由GS区与激酶共同组成的胞内区。该类受体的胞外区域较短,约占总长的20%,胞内区域较长。胞外近膜区域有10个半胱氨酸残基组成保守的富半胱氨酸盒,具有亲水性。胞内近膜区靠近蛋白激酶结构域有一段长为30个氨基酸左右的高度保守区域,包含TTSGGSGSGLP序列,富含丝氨酸和甘氨酸,称为GS结构域。TβRⅡ包括ActR-Ⅱ、ActR-ⅡB、Punt、TβR-Ⅱ、BMPR-Ⅱ等,其也由四个部分构成:信号肽、亲水胞外区、跨膜区和由激酶与富含Ser/Thr的短尾共同构成的胞内区,可发生自身的磷酸化作用。总体来说,TβRⅠ与TβRⅡ都是包含四部分结构的糖蛋白,胞外都含有10个左右的Cys残基,这些残基决定了胞外区域的折叠方式,胞内均有一个丝氨酸/苏氨酸激酶区。二者的主要的区别在于TβRⅠ的胞外区域短于TβRⅡ,而在胞内TβRⅠ含有高度保守的GS结构域,TβRⅡ在胞内无GS结构域,取而代之的是一个丝氨酸/苏氨酸的短尾。由于TβRⅠ不能进行自身的磷酸化,因此必须经过TβRⅡ激酶转磷酸化之后方具有活性,TβRⅠ GS结构域的丝氨酸/苏氨酸磷酸化是介导TGF-β信号通路的必需途径。
2.信号通路
TGF-β家族成员作为配体与相应的受体结合的方式主要有两种模式。一种结合形式的配体是BMP家族成员,另一种结合方式的配体包含TGF-β和活化素。BMP配体中的BMP2和BMP4等与胞外的受体TβRⅠ的结合区域具有很高的亲和力,而对于TβRⅡ则表现出较低的亲和力,但BMP与TβRⅠ结合后提升了与TβRⅡ的亲和力[119]。与BMP不同,TGF-β和活化素则与TβRⅡ表现出了更高的亲和力,而不会与TβRⅠ发生反应[120]。因此配体首先与TβRⅡ的胞外区紧密结合,该复合体可进一步与TβRⅠ结合,最终形成TβRⅠ-TGF-β-TβRⅡ异源四聚体,该四聚体在信号从细胞膜传入细胞核的过程中起到重要的作用。TβRⅡ胞外结构与TGF-β3复合物的结构研究显示二者的结合发生在远端[121]。目前尚未发现TβRⅢ进行激活细胞信号通路的途径,但TβRⅢ可起到清除和活化TGF-β的功能。其清除与活化功能与TβRⅢ所处的形式有关,当TβRⅢ处于溶解形式时,可抑制配体TGF-β结合到TβRⅡ上;当TβRⅢ与细胞膜结合时,其又可对TGF-β与TβRⅡ的结合起到促进作用[122]。
Sekelsky等[123]在1995年首先在果蝇中发现了补救Dpp分子基因dpp缺失突变的分子,将其命名为MAD,并证明其参与了TGF-β信号的胞内转导。随后在线虫以及脊椎动物中均发现该类蛋白,将其命名为Smad[124]。根据Smad蛋白的结构和功能,可将其分为3种亚族,分别为受调节的Smads(R-Smads)、共同通路型Smads(Co-Smads)和抑制性Smads(I-Smads)。其中R-Smads包含Smad1/2/3/5/8,决定了配体信号转导的特异性,其C末端具有特定的ser序列,TβRⅠ激酶将其磷酸化并激活后,可进入细胞核,进一步调节靶基因的表达。Smad2和Smad3参与TGF-β信号通路,而Smad1、Smad5和Smad8参与BMP信号通路。Co-Smads包含Smad4、Meden,TGF-β/Activin/Nodal和BMP/GDF/MIS信号通路均需要Smad4参与,方可将信号分子传递入核。Smad4在TGF-β信号通路中对基因表达的调控以及生长抑制等均是必不可少的重要转录因子[125]。I-Smad主要包含Smad6和Smad7,可与R-Smad竞争结合激活的TβRⅠ,在TGF-β信号通路中起到抑制作用[126-127]。
R-Smad和Co-Smad蛋白都是由500个氨基酸残基构成的,包含两个保守的机构区域:N末端的MH1以及C末端的MH2。而C末端的NH2结构区域的特征性丝氨酸序列(SXS motif)中的两个丝氨酸残基对R-Smad的活性起到介导作用。NH1和NH2结构区域均可与细胞核中的多种蛋白反应,从而影响转录。尽管在不同的smad中处于NH1和NH2间的连接序列各不相同,但它们都包含了多个可磷酸化位点,导致它们可与其它的信号通路之间发生交联作用;这些连接序列中也包含这一个PY motif,调节了与Smurfs间的特殊性相互作用[128]。
细胞内合成的TGF-β以无活性的状态分泌到细胞外,经过活化后方可与相关受体进行结合。由于R-Smad中的L3环上的NH2区域较Co-Smad的该区域带有更多的正电荷,导致磷酸化的TβRⅠ对R-Smad表现出了更大的亲和力[129]。而位于毗邻GS区域上的激酶受体区域的L45环与R-Smad具有特异性结合作用[130]。正是由于Smad L3环和L45环的差异决定了活化的TβRⅠ对受体Smad的选择性,因此TGF-β信号通路可以分为两个方向:TGF-β-活化素-nodal通路和骨形成蛋白-生长分化因子通路。TGF-β-活化素-nodal通路中,信号经过ALK4/5/7来激活下游的Smad2和Smad3;骨形成蛋白-生长分化因子通路中,信号通过ALK1/2/3/6来激活下游的Smad1、Smad5以及Smad8[128]。在TGF-β信号通路中存在一个“Smad循环”[131],活化的TβRⅠ可与细胞质中以同源寡聚体形式存在的Smad2和Smad3结合,将Smad2/3 C末端的SXS序列中的两个丝氨酸残基磷酸化而将其激活,随后与Smad4结合,形成异源六聚体进入细胞核中,从而作为转录因子单独或者与其它种类的DNA结合蛋白共同激活特定的靶基因完成转录。随后Smad复合物发生解离,而细胞核中的磷酸酶可将磷酸化的R-Smad脱去磷酸基,回复原来状态的R-Smad重新回到细胞质中,形成了“Smad循环”。
3.与肺纤维化的关系
TGF-β是研究最为透彻的一种致纤维化细胞因子,TGF-β可由肺中多种细胞分泌,其中包含肺泡巨噬细胞、中性粒细胞、激活的肺泡上皮细胞、内皮细胞、成纤维细胞以及肌成纤维细胞。TGF-β处于激活状态时,其具有包含趋化和促进增殖功能等的多效生长因子功能。TGF-β诱导了巨噬细胞和成纤维细胞的补充,并通过血小板生长因子(PDGF)的表达使成纤维细胞增生。TGF-β同样刺激了TNF-a,PDGF,IL-1b或IL-13等多种炎症因子和纤维化因子的表达,从而进一步强化和延续了纤维化反应。肺上皮细胞的反复受损可能是IPF中异常创伤修复程序发生的诱因[132]。上皮细胞受到破坏后,TGF-β1/Smad3介导的上皮中PAI-1上调导致血小板中富含纤维蛋白的凝血因子激活,以及强烈的抗纤维蛋白溶解的级联反应[133]。另有研究显示,使用siRNA致使PLI-1失活后可限制博来霉素小鼠模型肺纤维化的进展,提示通过PAI-1-siRNA限制肺泡凝聚可起到直接的抗纤维化作用[134]。体外研究显示,PAI-1-siRNA可抑制TGF-β在肺泡上皮细胞中介导的上皮间质转化,因此可将PAI-1作为IPF的潜在治疗位点。
多种因素的共同作用导致了在肺纤维化的发生,其中尤以细胞因子TGF-β的刺激作用为主,造成成纤维细胞不断的增殖并转型为肌成纤维细胞。目前认为TGF-β是导致肺纤维化最主要的细胞因子之一,其通过不同的信号路径,趋化炎症细胞和成纤维细胞,诱导了成纤维细胞向肌成纤维细胞的转化[135],也促进了细胞合成更多的TNF-α、IL-1等,亦可自我诱导性的扩大生物学效应,使细胞产生更多的TGF-β,并对胞外基质(ECM)起调节作用。研究显示,在使用博来霉素和环磷酰胺等多种可导致肺纤维化的药物之后,体内TGF-β含量出现升高现象,而使用TGF-β抗体进行阻断后,肺纤维化出现减轻现象[136]。
病理结果可以清晰显示IPF中的纤维化区域,该区域位于上皮下的大量激活的肌成纤维细胞位置,该位置紧靠受伤点或者ATⅡ增生细胞。这些激活的肌成纤维细胞可能在IPF时促进ECM的沉积过程中起到关键作用[137]。研究结果显示,纤维化集中区域的激活的肌成纤维细胞可能主要来源于以下几个方面:激活以及增生的原肺成纤维细胞,经由EMT过程而来的ATⅠ和ATⅡ细胞,由骨髓而来的成纤维细胞,或者是肺间隙组织周皮细胞[138]。因此,以成纤维细胞激活位点为治疗位点可作为IPF治疗的有效手段[137]。
六、展望
综上所述多种信号通路从多方面参肺纤维化的形成。但这是一个多因素的复杂过程,其具体调控及反馈调节机制、细胞因子网络的互作用、信号通路间的可能联系尚有待逐步揭示。进一步加强信号通路中炎症因子的研究,可能为延缓或阻断纤维化进程提供有益的尝试,并为临床治疗和早期预防肺纤维化提供理论依据。
1 Loewith R,Jacinto E,Wullschleger S,et al.Two TOR complexes,only one of which is rapamycin sensitive,have distinct roles in cell growth control[J].Mol Cell,2002,10(3):457-468.
2 Chiang GG,Abraham RT.Targeting the mTOR signaling network in cancer[J].Trends Mol Med,2007,13(10):433-442.
3 Hara K,Maruki Y,Long X,et al.Raptor,a binding partner of target of rapamycin (TOR),mediates TOR action[J].Cell,2002,110(2):177-189.
4 Sarbassov DD,Ali SM,Kim DH,et al.Rictor,a novel binding partner of mTOR,defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton[J].Curr Biol,2004,14(14):1296-1302.
5 Frias MA,Thoreen CC,Jaffe JD,et al.mSin1 is necessary for Akt/PKB phosphorylation,and its isoforms define three distinct mTORC2s[J].Curr Biol,2006,16(18):1865-1870.
6 Pearce LR,Huang X,Boudeau J,et al.Identification of Protor as a novel Rictor-binding component of mTOR complex-2[J].Biochem J,2007,405(405):513-522.
7 Sarbassov DD,Ali SM,Sengupta S,et al.Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB[J].Mol Cell,2006,22(2):159-168.
8 Guertin DA,Sabatini DM.Defining the role of mTOR in cancer[J].Cancer Cell,2007,12(1):9-22.
9 Jacinto E,Loewith R,Schmidt A,et al.Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive[J].Nat Cell Biol,2004,6(11):1122-1128.
10 Choo AY,Yoon SO,Kim SG,et al.Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation[J].Proc Natl Acad Sci USA,2008,105(45):17414-17419.
11 Gwinn DM,Shackelford DB,Egan DF,et al.AMPK phosphorylation of raptor mediates a metabolic checkpoint[J].Mol Cell,2008,30(2):214-226.
12 Inoki K,Zhu T,Guan KL.TSC2 mediates cellular energy response to control cell growth and survival[J].Cell,2003,115(5):577-590.
13 Gingras AC,Raught B,Sonenberg N.Regulation of translation initiation by FRAP/mTOR[J].Genes Dev,2001,15(7):807-826.
14 Shimada H,Hirai K,Simamura E,et al.Paraquat toxicity induced by voltage-dependent anion channel 1 acts as an NADH-dependent oxidoreductase[J].J Biol Chem,2009,284(42):28642-28649.
15 梁敏锐.肺纤维化过程中肌成纤维细胞活化的调控机制及干预研究[D].上海:复旦大学,2013.
16 邵雪.雷帕霉素治疗小鼠百草枯中毒致肺纤维化的作用及机制研究[D].杭州:浙江大学,2015.
17 万安霞.加减补肺汤对博来霉素诱导小鼠肺纤维化干预作用的实验研究[D].武汉:湖北中医药大学,2015.
18 王密琳.煤工尘肺患者肺巨噬细胞自噬活动研究[D].唐山:河北联合大学,2014.
19 Gui YS,Wang L,Tian X,et al.mTOR Overactivation and Compromised Autophagy in the Pathogenesis of Pulmonary Fibrosis[J].PLoS One,2015,10(9):e0138625.
20 Patel AS,Lin L,Geyer A,et al.Autophagy in idiopathic pulmonary fibrosis[J].PloS one,2012,7(7):e41394.
21 White ES,Sagana RL,Booth AJ,et al.Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway[J].Exp Cell Res,2010,316(16):2644-2653.
22 Morgan TH.The theory of the gene[J].American Naturalist,1917,51(609):513-544.
23 Artavanis-Tsakonas S,Matsuno K,Fortini ME.Notch signaling[J].Science,1995,268(5208):225-232.
24 Koch U,Radtke F.Chapter Thirteen - Notch Signaling in Solid Tumors[J].Curr Topics Dev Biol,2010,92(6):411-455.
25 Miele L.Notch signaling[J].Clin Cancer Res,2006,12(12):1074-1079.
26 Carlson ME,Conboy IM.Regulating the Notch pathway in embryonic,adult and old stem cells[J].Curr Opin Pharmacol,2007,7(3):303-309.
27 Fiúza UM,Arias AM.Cell and molecular biology of Notch[J].The Journal of endocrinology,2007,194(3):459-474.
28 Jarriault S,Brou C,Logeat F,et al.Signalling downstream of activated mammalian Notch[J].Nature,1995,377(6547):355-358.
29 Nam Y,Aster JC,Blacklow SC.Notch signaling as a therapeutic target[J].Current Opinion in Chemical Biology,2002,6(4):501-509.
30 Takasugi N,Tomita T,Hayashi I,et al.The role of presenilin cofactors in the gamma-secretase complex[J].Nature,2003,422(6930):438-441.
31 Kovall RA.Structures of CSL,Notch and Mastermind proteins:piecing together an active transcription complex[J].Curr Opin Struct Biol,2007,17(1):117-127.
32 Martinez Arias A,Zecchini V,Brennan K.CSL-independent Notch signalling:a checkpoint in cell fate decisions during development?[J].Curr Opin Genet Dev,2002,12(5):524-533.
33 Yamamoto N,Yamamoto S,Inagaki F,et al.Role of Deltex-1 as a transcriptional regulator downstream of the Notch receptor[J].J Biol Chem,2001,276(48):45031-45040.
34 Aoyagi-Ikeda K,Maeno T,Matsui H,et al.Notch induces myofibroblast differentiation of alveolar epithelial cells via transforming growth factor-{beta}-Smad3 pathway[J].Am J Respir Cell Mol Biol,2011,45(1):136-144.
35 Plantier L,Crestani B,Wert SE,et al.Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis[J].Thorax,2011,66(8):651-657.
36 Kavian N,Servettaz A,Mongaret C,et al.Targeting ADAM-17/notch signaling abrogates the development of systemic sclerosis in a murine model[J].Arthritis Rheum,2010,62(11):3477-3487.
37 Ostroukhova M,Qi Z,Oriss TB,et al.Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-beta[J].J Clin Invest,2006,116(4):996-1004.
38 Fitau J,Boulday G,Coulon F,et al.The adaptor molecule Lnk negatively regulates tumor necrosis factor-alpha-dependent VCAM-1 expression in endothelial cells through inhibition of the ERK1 and -2 pathways[J].J Biol Chem,2006,281(29):20148-20159.
39 Liu T,Hu B,Choi YY,et al.Notch1 signaling in FIZZ1 induction of myofibroblast differentiation[J].Am J Pathol,2009,174(5):1745-1755.
40 Piggott K,Deng J,Warrington K,et al.Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis[J].Circulation,2011,123(3):309-318.
41 Wang Q,Li DC,Li ZF,et al.Upregulation of miR-27a contributes to the malignant transformation of human bronchial epithelial cells induced by SV40 small T antigen[J].Oncogene,2011,30(36):3875-3886.
42 Nusse R,Varmus HE.Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome[J]Cell,1982,31(1):99-109.
43 Nusse R,Brown A,Papkoff J,et al.A new nomenclature for int-1 and related genes:the Wnt gene family[J].Cell,1991,64(2):231.
44 Gordon MD,Nusse R.Wnt signaling:multiple pathways,multiple receptors,and multiple transcription factors[J].J Biol Chem,2006,281(32):22429-22433.
45 Van De Schans VA,Smits JF,Blankesteijn WM.The Wnt/frizzled pathway in cardiovascular development and disease:friend or foe?[J].Eur J Pharmacol,2008,585(2-3):338-345.
46 Niehrs C.The complex world of WNT receptor signalling[J].Nat Rev Mol Cell Biol,2012,13(12):767-779.
47 Kikuchi A,Yamamoto H,Kishida S.Multiplicity of the interactions of Wnt proteins and their receptors[J].Cell Signal,2007,19(4):659-671.
48 Xing Y,Clements W K,Kimelman D,et al.Crystal structure of a beta-catenin/axin complex suggests a mechanism for the beta-catenin destruction complex[J].Genes Dev,2003,17(22):2753-2764.
49 Willert K,Nusse R.Beta-catenin:a key mediator of Wnt signaling[J].Current opinion in genetics & development,1998,8(1):95-102.
50 Logan CY,Nusse R.The Wnt signaling pathway in development and disease[J].Annu Rev Cell Dev Biol,2004,20:781-810.
51 Zeng X,Tamai K,Doble B,et al.A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation[J].Nature,2005,438(7069):873-877.
52 Li VS,Ng SS,Boersema PJ,et al.Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex[J].Cell,2012,149(6):1245-1256.
53 Van Amerongen R,Nusse R.Towards an integrated view of Wnt signaling in development[J].Development,2009,136(19):3205-3214.
54 Kulikova KV,Posviatenko AV,Gnuchev NV,et al.[Nuclear beta-catenin localization is not sufficient for canonical Wnt signaling activation in human meianoma cell lines][J].Mol Biol(Mosk),2011,45(5):884-891.
55 Graham TA,Weaver C,Mao F,et al.Crystal structure of a beta-catenin/Tcf complex[J].Cell,2000,103(6):885-896.
56 Mosimann C,Hausmann G,Basler K.Beta-catenin hits chromatin:regulation of Wnt target gene activation[J].Nat Rev Mol Cell Biol,2009,10(4):276-286.
57 Kim TH,Kim SH,Seo JY,et al.Blockade of the Wnt/beta-catenin pathway attenuates bleomycin-induced pulmonary fibrosis[J].Tohoku J Exp Med,2011,223(1):45-54.
58 Raghu G,Rochwerg B,Zhang Y,et al.An Official ATS/ERS/JRS/ALAT clinical practice guideline:treatment of idiopathic pulmonary fibrosis.An update of the 2011 clinical practice guideline[J].Am J Respir Crit Care Med,2015,192(2):e3-19.
59 Park S,Lee EJ.Recent advances in idiopathic pulmonary fibrosis[J].Tuberc Respir Dis(Seoul),2013,74(1):1-6.
60 Konigshoff M,Kramer M,Balsara N,et al.WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis[J].J Clin Invest,2009,119(4):772-787.
61 Chilosi M,Poletti V,Zamò A,et al.Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis[J].Am J Pathol,2003,162(5):1495-1502.
62 Uhal BD,Joshi I,Hughes WF,et al.Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung[J].Am J Physiol,1998,275(6Pt1):L1192-L1199.
63 Uhal BD,Joshi I,True AL,et al.Fibroblasts isolated after fibrotic lung injury induce apoptosis of alveolar epithelial cells in vitro[J].Am J Physiol,1995,269(6 Pt 1):L819-L828.
64 Selman M,Ruiz V,Cabrera S,et al.TIMP-1,-2,-3,and -4 in idiopathic pulmonary fibrosis.A prevailing nondegradative lung microenvironment?[J].Am J Physiol Lung Cell Mol Physiol,2000,279(3):L562-574.
65 Kaminski N,Rosas IO.Gene expression profiling as a window into idiopathic pulmonary fibrosis pathogenesis:can we identify the right target genes?[J].Proc Am Thorac Soc,2006,3(4):339-344.
66 Liu L,Carron B,Yee HT,et al.Wnt pathway in pulmonary fibrosis in the bleomycin mouse model[J].J Environ Pathol Toxicol Oncol,2009,28(2):99-108.
67 Königshoff M,Balsara N,Pfaff E M,et al.Functional Wnt signaling is increased in idiopathic pulmonary fibrosis[J].PLoS One,2008,3(5):e2142.
68 Pfaff EM,Becker S,Gunther A,et al.Dickkopf proteins influence lung epithelial cell proliferation in idiopathic pulmonary fibrosis[J].Eur Respir J,2011,37(1):79-87.
69 Lam AP,Flozak AS,Russell S,et al.Nuclear β-Catenin Is Increased in Systemic Sclerosis Pulmonary Fibrosis and Promotes Lung Fibroblast Migration and Proliferation [J].Am J Respir Cell Mol Biol,2011,45(5):915-922.
70 Nusslein-Volhard C,Wieschaus EF.Mutations affecting segment number and polarity in Drosophila[J].Nature,1980,287(5785):795-801.
71 Mohler J.Requirements for hedgehog,a segmental polarity gene,in patterning larval and adult cuticle of Drosophila[J].Genetics,1988,120(4):1061-1072.
72 Forbes AJ,Nakano Y,Taylor AM,et al.Genetic analysis of hedgehog signalling in the Drosophila embryo[J].Dev Suppl,1993:115-124.
73 Ingham PW,Mcmahon AP.Hedgehog signaling in animal development:paradigms and principles[J].Genes Dev,2001,15(23):3059-3087.
74 Ingham PW,Nakano Y,Seger C.Mechanisms and functions of Hedgehog signalling across the metazoa[J].Nat Rev Genet,2011,12(6):393-406.
75 Taipale J,Beachy PA.The Hedgehog and Wnt signalling pathways in cancer[J].Nature,2001,411(6835):349-354.
76 Pan Y,Wang B.A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome[J].J Biol Chem,2007,282(15):10846-10852.
77 Porter JA,Von Kessler DP,Ekker SC,et al.The product of hedgehog autoproteolytic cleavage active in local and long-range signalling[J].Nature,1995,374(6520):363-366.
78 Porter JA,Ekker SC,Park WJ,et al.Hedgehog patterning activity:role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain[J].Cell,1996,86(1):21-34.
79 Lee JJ,Ekker SC,Von Kessler DP,et al.Autoproteolysis in hedgehog protein biogenesis[J].Science,1994,266(5190):1528-1537.
80 Porter JA,Young KE,Beachy PA.Cholesterol modification of hedgehog signaling proteins in animal development[J].Science,1996,274(5285):255-259.
81 Chen X,Tukachinsky H,Huang CH,et al.Processing and turnover of the Hedgehog protein in the endoplasmic reticulum[J].J Cell Biol,2011,192(5):825-838.
82 Buglino JA,Resh MD.Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog[J].J Biol Chem,2008,283(32):22076-22088.
83 Zhu AJ,Scott MP.Incredible journey:how do developmental signals travel through tissue?[J].Genes Dev,2004,18(24):2985-2997.
84 Burke R,Nellen D,Bellotto M,et al.Dispatched,a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells[J].Cell,1999,99(7):803-815.
85 Gallet A.Hedgehog morphogen:from secretion to reception[J].Trends Cell Biol,2011,21(4):238-246.
86 Chen MH,Li YJ,Kawakami T,et al.Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates[J].Genes Dev,2004,18(6):641-659.
87 Eugster C,Panáková D,Mahmoud A,et al.Lipoprotein-heparan sulfate interactions in the Hh pathway[J].Dev Cell,2007,13(1):57-71.
88 Dierker T,Dreier R,Petersen A,et al.Heparan sulfate-modulated,metalloprotease-mediated sonic hedgehog release from producing cells[J].J Biol Chem,2009,284(12):8013-8022.
89 Guerrero I,Chiang C.A conserved mechanism of Hedgehog gradient formation by lipid modifications[J].Trends Cell Biol,2007,17(1):1-5.
90 Pepinsky RB,Zeng C,Wen D,et al.Identification of a palmitic acid-modified form of human Sonic hedgehog[J].J Biol Chem,1998,273(22):14037-14045.
91 Chamoun Z,Mann RK,Nellen D,et al.Skinny hedgehog,an acyltransferase required for palmitoylation and activity of the hedgehog signal[J].Science,2001,293(5537):2080-2084.
92 Routh MD,Zalucki Y,Su CC,et al.Efflux pumps of the resistance-nodulation-division family:a perspective of their structure,function,and regulation in gram-negative bacteria[J].Adv Enzymol Relat Areas Mol Biol,2011,77:109-146.
93 Allen BL,Song JY,Izzi L,et al.Overlapping roles and collective requirement for the coreceptors GAS1,CDO,and BOC in SHH pathway function[J].Dev Cell,2011,20(6):775-787.
94 Taipale J,Cooper MK,Maiti T,et al.Patched acts catalytically to suppress the activity of Smoothened[J].Nature,2002,418(6900):892-897.
95 Rohatgi R,Milenkovic L,Scott MP.Patched1 regulates hedgehog signaling at the primary cilium[J].Science,2007,317(5836):372-376.
96 Wang Y,Zhou Z,Walsh CT,et al.Selective translocation of intracellular Smoothened to the primary cilium in response to Hedgehog pathway modulation[J].Proc Natl Acad Sci U S A,2009,106(8):2623-2628.
97 Chen JK,Taipale J,Young KE,et al.Small molecule modulation of Smoothened activity[J].Proc Natl Acad Sci U S A,2002,99(22):14071-14076.
98 Chen Y,Sasai N,Ma G,et al.Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened[J].PLoS Biol,2011,9(6):e1001083.
99 Milenkovic L,Scott MP Rohatgi R.Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium[J].J Cell Biol,2009,187(3):365-374.
100 Stewart GA,Hoyne GF,Ahmad SA,et al.Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes[J].J Pathol,2003,199(4):488-495.
101 Kasper M,Haroske G.Alterations in the alveolar epithelium after injury leading to pulmonary fibrosis[J].Histol Histopathol,1996,11(2):463-483.
102 Khalil N,O’connor RN,Flanders KC,et al.TGF-beta 1,but not TGF-beta 2 or TGF-beta 3,is differentially present in epithelial cells of advanced pulmonary fibrosis:an immunohistochemical study[J].Am J Respir Cell Mol Biol,1996,14(2):131-138.
103 Levine D,Rockey DC,Milner TA,et al.Expression of the integrin alpha8beta1 during pulmonary and hepatic fibrosis[J].Am J Pathol,2000,156(6):1927-1935.
104 Bellusci S,Furuta Y,Rush MG,et al.Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis[J].Development,1997,124(1):53-63.
105 Weaver M,Batts L,Hogan BL.Tissue interactions pattern the mesenchyme of the embryonic mouse lung[J].Dev Biol,2003,258(1):169-184.
106 Liu L,Kugler MC,Loomis CA,et al.Hedgehog signaling in neonatal and adult lung[J].Am J Respir Cell Mol Biol,2013,48(6):703-710.
107 Coon DR,Roberts DJ,Loscertales M,et al.Differential epithelial expression of SHH and FOXF1 in usual and nonspecific interstitial pneumonia[J].Exp Mol Pathol,2006,80(2):119-123.
108 Bolanos AL,Milla CM,Lira JC,et al.Role of Sonic Hedgehog in idiopathic pulmonary fibrosis[J].Am J Physiol Lung Cell Mol Physiol,2012,303(11):L978-L990.
109 Horn A,Palumbo K,Cordazzo C,et al.Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis[J].Arthritis Rheum,2012,64(8):2724-2733.
110 Tuder RM.A pathologist’s approach to interstitial lung disease[J].Curr Opin Pulm Med,1996,2(5):357-363.
111 Katoh M,Katoh M.Human FOX gene family(Review)[J].Int J Oncol,2004,25(5):1495-1500.
112 Tseng HT,Shah R,Jamrich M.Function and regulation of FoxF1 during Xenopus gut development[J].Development,2004,131(15):3637-3647.
113 Roberts AB,Lamb LC,Newton DL,et al.Transforming growth factors:isolation of polypeptides from virally and chemically transformed cells by acid/ethanol extraction[J].Proc Natl Acad Sci U S A,1980,77(6):3494-3498.
114 De Larco JE,Todaro GJ.Growth factors from murine sarcoma virus-transformed cells[J].Proc Natl Acad Sci U S A,1978,75(8):4001-4005.
115 Brodin G,Ten Dijke P,Funa K,et al.Increased smad expression and activation are associated with apoptosis in normal and malignant prostate after castration[J].Cancer Res,1999,59(11):2731-2738.
116 Wu MY,Hill CS.Tgf-beta superfamily signaling in embryonic development and homeostasis[J].Dev Cell,2009,16(3):329-343.
117 牛秀珑,王越,梁国庆.转化生长因子β的研究进展[J].医学综述,2004,10(4):209-210.
118 Liu T,Feng XH.Regulation of TGF-beta signalling by protein phosphatases[J].Biochem J,2010,430(2):191-198.
119 Kirsch T,Sebald W,Dreyer MK.Crystal structure of the BMP-2-BRIA ectodomain complex[J].Nat Struct Biol,2000,7(6):492-496.
120 Massague J.TGF-beta signal transduction[J].Annu Rev Biochem,1998,67:753-791.
121 Hart PJ,Deep S,Taylor AB,et al.Crystal structure of the human TbetaR2 ectodomain——TGF-beta3 complex[J].Nat Struct Biol,2002,9(3):203-208.
122 Taylor AW.Review of the activation of TGF-beta in immunity[J].Journal of leukocyte biology,2009,85(1):29-33.
123 Sekelsky JJ,Newfeld SJ,Raftery LA,et al.Genetic characterization and cloning of mothers against dpp,a gene required for decapentaplegic function in Drosophila melanogaster[J].Genetics,1995,139(3):1347-1358.
124 Derynck R,Gelbart WM,Harland RM,et al.Nomenclature:vertebrate mediators of TGFbeta family signals[J].Cell,1996,87(2):173.
125 Dennler S,Goumans M J,Ten Dijke P.Transforming growth factor beta signal transduction[J].J Leukoc Biol,2002,71(5):731-740.
126 Imamura T,Takase M,Nishihara A,et al.Smad6 inhibits signalling by the TGF-beta superfamily[J].Nature,1997,389(6651):622-626.
127 Nakao A,Afrakhte M,Moren A,et al.Identification of Smad7,a TGFbeta-inducible antagonist of TGF-beta signalling[J].Nature,1997,389(6651):631-635.
128 Miyazawa K,Shinozaki M,Hara T,et al.Two major Smad pathways in TGF-beta superfamily signalling[J].Genes Cells,2002,7(12):1191-1204.
129 Wu G,Chen YG,Ozdamar B,et al.Structural basis of Smad2 recognition by the Smad anchor for receptor activation[J].Science,2000,287(5450):92-97.
130 Chen YG,Hata A,Lo R S,et al.Determinants of specificity in TGF-beta signal transduction[J].Genes Dev,1998,12(14):2144-5212.
131 Lin X,Duan X,Liang YY,et al.PPM1A Functions as a Smad Phosphatase to Terminate TGFbeta Signaling[J].Cell,2016,166(6):1597.
132 Wilson MS,Wynn TA.Pulmonary fibrosis:pathogenesis,etiology and regulation[J].Mucosal Immunol,2009,2(2):103-121.
133 Dennler S,Itoh S,Vivien D,et al.Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene[J].Embo j,1998,17(11):3091-3100.
134 Senoo T,Hattori N,Tanimoto T,et al.Suppression of plasminogen activator inhibitor-1 by RNA interference attenuates pulmonary fibrosis[J].Thorax,2010,65(4):334-340.
135 Piek E,Heldin CH,Ten Dijke P.Specificity,diversity,and regulation in TGF-beta superfamily signaling[J].FASEB J,1999,13(15):2105-2124.
136 Giri SN,Hyde DM,Hollinger MA.Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice[J].Thorax,1993,48(10):959-966.
137 Eickelberg O,Laurent GJ.The quest for the initial lesion in idiopathic pulmonary fibrosis:gene expression differences in IPF fibroblasts[J].Am J Respir Cell Mol Biol,2010,42(1):1-2.
138 Scotton CJ,Chambers RC.Molecular targets in pulmonary fibrosis:the myofibroblast in focus[J].Chest,2007,132(4):1311-1321.
10.3877/cma.j.issn.2095-9133.2017.04.011
2012年国家临床重点专科建设项目(2012650);泰山学者建设工程专项经费项目(ts20130911);山东省重点研发计划项目(2015GSF118038,2016GSF201041);山东大学横向合作计划项目(11671405,11671423,11671511);山东省医药卫生科技发展计划项目(2014WS0150 ,2013BJYB12 );山东省医药卫生重点实验室(鲁卫科教国合字[2013]49号)
250012 山东济南,山东大学齐鲁医院急诊科中毒与职业病科1;264200 山东威海,山东省威海市立医院2;255036 山东淄博市中心医院3;230001 安徽合肥,安徽省立医院4
菅向东,Email:jianxiangdongvip@163.com
2017-07-01)
(本文编辑:楚鹰)
宋玲莉,李海,陶晓根,等.信号转导通路与肺纤维化关系的研究进展[J/CD].中华卫生应急电子杂志,2017,3(4):242-252.
