槐角提取物中槐角苷的含量测定研究*
2017-12-27刘金香潘五九王伟明
刘金香 潘五九 王伟明 董 坤 姚 琳
(黑龙江省中医药科学院·哈尔滨 150036)
·方药研究·
槐角提取物中槐角苷的含量测定研究*
刘金香 潘五九**王伟明 董 坤 姚 琳
(黑龙江省中医药科学院·哈尔滨 150036)
目的:建立槐角提取物中槐角苷的含量测定方法。方法:采用Diamonsil C18色谱柱(250mm×4.6mm,5μm);流动相:乙腈-0.07%磷酸水溶液(25:75);流速1.0mL/min;检测波长260nm;柱温:30℃;进样量:10μL。结果:槐角苷进样浓度在1.502~300.400μg/mL范围内与峰面积线性关系良好,回归方程为:Y=4807X+2870(r=0.9999),平均加样回收率为101.50%,RSD为1.45%。结论:所建立的方法简单可行,重复性好,可用于槐角提取物中槐角苷的含量测定。
槐角 提取物 槐角苷 含量测定 高效液相色谱法
槐角为豆科植物槐(Sophorajaponica L.)的干燥成熟果实,性苦、寒 ,具有清热泻火,凉血止血功效[1]。槐角中含多种黄酮类和异黄酮类化合物,其中槐角苷是槐角的主要活性成分之一[2]。现代研究表明槐角苷具有抗炎、抑制免疫、抗生育和雌激素样等作用[3-4]。我院通过前期的试验研究,分离纯化出槐角提取物,其主要成分为槐角苷。目前,对于槐角药材中槐角苷[5-7]含量测定研究较多,而槐角提取物中的槐角苷的含量测定研究尚未见报道,为有效控制槐角提取物质量,本实验以所在科研课题为支撑,通过HPLC方法测定槐角提取物中槐角苷的含量,为槐角提取物的开发利用提供实验依据。
1 材料
岛津2010LC高效液相色谱仪,SPD-10AVvp检测器(岛津);Sartorius BSA224S-CW电子天平(赛多利斯科学仪器北京有限公司),Sartorius BP211D电子天平(上海精密仪器有限公司);KQ-300DB型数控超声波清洗器(昆山市超声仪器有限公司)。槐角苷对照品(中国食品药品检定研究院购买,批号为111695—200501);槐角提取物(自制,批号为20110901,20010902,20110903),乙腈、甲醇、磷酸为色谱级,其余试剂均为分析纯,水为哇哈哈纯净水。
2 方法及结果
2.1 色谱条件
色谱柱:Diamonsil C18(250mm×4.6mm,5µm);流动相:乙腈–0.07%磷酸水溶液(25:75);流速:1.0mL/min;波长:260nm;柱温:30℃;进样量:10µL。理论板数按槐角苷峰计算应不低于5000。样品中槐角苷峰与相近峰达到基线分离,分离度良好。
2.2 溶液的制备
2.2.1 对照品溶液的制备 精密称取槐角苷对照品适量,加甲醇制成每1ml含300.400µg的溶液,作为对照品储备液。精密吸取该储备液1ml,置于10ml容量瓶中,加入甲醇定容至刻度,摇匀,得到对照品溶液。
2.2.2 供试品溶液的制备 取本品粉末约20mg,精密称定,置50ml量瓶中,加入70%乙醇40ml,超声处理(功率300W,频率40kHz)15分钟,放冷,加70%乙醇至刻度,摇匀,滤过,取续滤液1ml置10ml量瓶中,加70%乙醇至刻度,摇匀,即得。
2.3 专属性试验
取上述对照品溶液和供试品溶液,按照2.1项下色谱条件,分别进样10ul,得到对照品、供试品色谱图,见图1、图2。样品中槐角苷峰与相近峰达到基线分离,分离度良好。

图1 槐角苷对照品色谱图

图2 供试品溶液色谱图
2.4 方法学考察
2.4.1 线性关系的考察 分别精密吸取上述对照品储备液 0.25,0.50,2.50,5.00,12.50ml置 50ml量瓶中,加甲醇至刻度,摇匀,制得系列标准溶液,依次精密吸取系列标准溶液和对照品储备液各10μl,按上述色谱条件测定峰面积,以槐角苷峰面积作为纵坐标,以槐角苷质量浓度(μg/mL)为横坐标进行线性回归,得到回归方程为Y=4807X+2870,r=0.9999,表明槐角苷质量浓度在1.502~300.4μg/mL范围内线性关系良好。
2.4.2 仪器的精密度试验 取同一份槐角苷对照品溶液适量,按上述色谱条件,连续进样6次,测定槐角苷峰面积,计算相对标准偏差(RSD)值为0.13%,结果表明该方法下仪器的精密度较好。
2.4.3 稳定性试验 按2.2.2项下方法制备供试液,分别于0、1、2、4、6、8进样,测定槐角苷峰面积,计算其峰面积相对标准偏差(RSD)为0.23%,表明样品溶液在8h内稳定性良好。
2.4.4 重复性试验 同一批号供试品,按2.2.2项下方法制备6份供试品溶液,按2.1项下色谱条件依次操作,测定槐角苷峰面积,其RSD为0.49%,结果表明该方法重现性较好。
2.4.5 加样回收率试验 取已知槐角苷含量的供试品约10mg(批号20110901),共6份,精密称定,分别精准加入等量的槐角苷对照品,按2.2.2项下方法制备槐角提取物供试品溶液,按2.1项下色谱条件对样品进行测定,计算平均回收率及RSD。测得结果见如下表1。
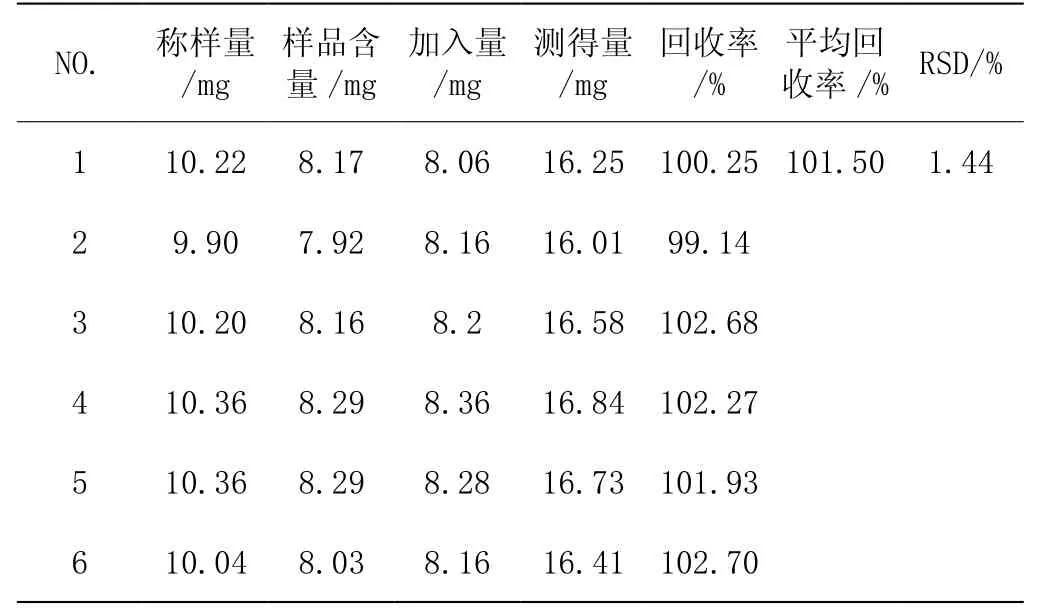
表1 槐角提取物中槐角苷加样回收率试验结果(n=6)
2.5 样品测定
三批样品,按照2.2.2项下方法,每个批号平行制备两份供试品溶液,按2.1项下色谱条件,每份供试品溶液重复测定两次。结果样品中槐角苷的含量的平均值分别为79.06%,79.59%,80.23%。
2.6 耐用性试验
考不同流速(0.8,0.9,1.0,1.1,1.2mL/min),不同柱温(25,30,35,40℃),不同色谱柱(Diamonsil C18,Agilent Excend C18)下色谱峰分离度和柱效的变化,结果,分离度和柱效无明显变化,表明此方法的耐用性良好。
3 结果与讨论
3.1 供试品制备方法
参考相关文献[8]中供试品溶液制备方法,分别对提取溶剂(甲醇,不同浓度乙醇)、超声提取时间(10、15、20、30、45min)进行考察,确定最优提取条件为70%乙醇,超声提取15min。
3.2 流动相的选择
参考相关文献[1,9,10]中槐角苷含量测定方法,考察了甲醇-乙腈-0.07%磷酸水,甲醇-0.07%磷酸水,乙腈-0.07%磷酸水系统,经多次实验发现,乙腈-0.07%磷酸水(25:75),目标峰保留时间适中,且色谱峰峰形对称性良好,因此,确定流动相组成及比例为乙腈-0.07%磷酸水(25:75)。
综上所述,本实验所建立槐角提取物的含量测定方法,简单易行,结果准确,重复性好,能够有效控制槐角提取物的质量,并为该提取物的质量标准的建立提供科学依据。
[1] 国家药典委员会.中国华人民共和国药典[M].一部.北京:化学工业出版社,2015:355-356.
[2] 常新全,丁丽霞.中药活性成分分析手册[M].学苑出版社,2002.
[3] Zhao C Q ,et al.Traditional Chinese medicine active ingredient analysis,beijing National Academy Press[M],2002: 22091.
[4] 瞿成权,孙齐,周建宏等.槐角苷对雌性小鼠的抗生育作用研究[J]. 四川动物,2014,33(04):558-562.
[5] 韦华梅,李飞,王羽,王剑波.HPLC法测定不同来源槐角中槐角苷含量[J]. 现代生物医学进展,2012,12(16):3170-3173.
[6] 杨子华,袁继承. 不同产地蜜槐角中槐角苷的含量测定[J].山东中医杂志,2012,31(09):671-673.
[7] 史亚军,唐恬. 不同产地槐角中槐角苷含量测定研究[J]. 现代中医药,2016,36(06):108-110.
[8] 孟庆杰,潘五九,王伟明,刘姗姗,付磊.高效液相色谱法测定槐角总黄酮水解产物中染料木素和槐角苷含量[J]. 中国药业,2014,23(19):40-42.
[9] 于西泉,肖华,李少华.健尔胶囊的质量标准研究和急性毒性试验[J].药物研究,2009,18(23):16-17.
[10] 房敏峰, 曲欢欢, 文颂华,等. 槐角不同炮制品中槐角苷的含量测定[J]. 中药材, 2007, 30(1):24-25.
2013哈尔滨市科技攻关项目(2013AA3BS040)
** 通讯作者
