不同发育阶段斯氏副柔线虫比较转录组学分析
2017-12-18王文龙冯陈晨红梅岳建伟呼和巴特尔刘春霞
王文龙,冯陈晨,红梅,岳建伟,呼和巴特尔,刘春霞
不同发育阶段斯氏副柔线虫比较转录组学分析
王文龙1,冯陈晨1,红梅1,岳建伟1,呼和巴特尔1,刘春霞2
(1内蒙古农业大学兽医学院/农业部动物疾病临床诊疗技术重点实验室,呼和浩特 010018;2内蒙古农业大学生命科学学院,呼和浩特 010018)
探明不同发育阶段骆驼斯氏副柔线虫的转录组差异,了解不同发育阶段虫体在功能分类和代谢通路等方面的生物学特征,挖掘生长发育相关功能基因,丰富寄生性线虫转录组学信息。采用Illumina HiSeq2000TM高通量测序技术对斯氏副柔线虫虫卵、第三期幼虫和雌虫进行转录组测序,构建3个样本的cDNA文库,评估建库质量;利用Trinity软件对所得序列进行De novo组装及组装效率评估;之后对获得的有效序列进行功能注释及相关生物信息学分析。测序及组装后虫卵、第三期幼虫和雌虫分别获得47 717、76 342和54 624个Unigenes,在虫卵和第三期幼虫比对中共有33 579个差异表达基因,其中表达上调的基因有20 477个,表达下调的基因有13 102个;在雌虫和第三期幼虫比对中共有32 199个差异表达基因,其中表达上调的基因有9 293个,表达下调的基因有22 906个。将成对比较的差异表达基因分别进行GO功能分类,其中虫卵和第三期幼虫比对中有6 617、3 891和8 755个Unigenes,雌虫和第三期幼虫比对中有7 043、3 686和10 177个Unigenes分别注释到生物过程、细胞组成和分子功能三大类中;通过KEGG pathway数据库分析,虫卵和第三期幼虫比对中有6 521个差异表达基因参与到251条通路中,显著富集MAPK信号通路、Wnt信号通路和氧化磷酸化等通路,雌虫和第三期幼虫比对中有6 528个参与到251条通路中,显著富集新陈代谢通路、DNA复制和细胞周期等通路。差异表达基因功能聚类分析中,雌虫和虫卵在调控生长速率和生殖发育等方面高表达;第三期幼虫在防御机制和糖类代谢等方面高表达;且3个发育阶段均在胚胎发育,胚后发育中富集高表达,其中在胚胎发育和胚后发育中分别有242和202个相关功能基因在3个发育阶段都表达,这些基因可能在胚胎(后)发育过程中起核心作用。此外,获得了9种异时性基因(如:LIN-28、LIN-14和RHEB-1等)、48个核激素受体(NHRs),包括NHR-49,NHR-48,NHR-40,NHR-1等以及36个锌金属蛋白酶(NAS),包括NAS-36、NAS-33和NAS-14等,并对其在斯氏副柔线虫虫卵、第三期幼虫和雌虫中的富集程度进行分析,发现这些基因在维持线虫正常生长发育过程中发挥关键作用,利用RNA-seq技术对3个发育阶段的斯氏副柔线虫进行测序和生物信息学分析,研究了不同发育阶段虫体的差异表达基因在GO功能分类、KEGG代谢通路和基因功能聚类等方面的生物学特性,筛选出多种异时性基因和发育相关重要基因,为后续开展斯氏副柔线虫功能基因组学研究,虫体与宿主互作、致病机制、免疫逃避等研究提供了理论依据。
斯氏副柔线虫;转录组;差异表达基因;发育相关基因
0 引言
【研究意义】骆驼斯氏副柔线虫()属于旋尾目、副柔属[1-2],是一种寄生于偶蹄反刍动物真胃的吸血性线虫,骆驼是其最适宜终末宿主。大量感染斯氏副柔线虫后,可引起骆驼腹泻,贫血甚至死亡。据资料显示,在中国内蒙古巴彦淖尔市双峰驼斯氏副柔线虫的感染率高达91.7%,感染强度最高可达1 315条,严重威胁骆驼的健康[3]。2009年,赵治国等在吸血蝇体内发现斯氏副柔线虫的第三期幼虫,并首次明确了截脉角蝇和西方角蝇是骆驼斯氏副柔线虫病的传播媒介[3]。但是有关骆驼斯氏副柔线虫在传播媒介与终末宿主体内不同发育阶段虫体的代谢水平差异、发育相关重要基因表达及致病机理等方面的研究并未见报道,严重阻碍了骆驼斯氏副柔线虫病的防控与治疗。因此,开展骆驼斯氏副柔线虫不同发育阶段虫体的差异表达基因研究对于从根本上解决斯氏副柔线虫对骆驼的危害具有十分重要的意义。【前人研究进展】随着高通量测序技术的快速发展,转录组测序已被广泛应用到不同发育阶段生物个体的基因差异表达研究中。目前,寄生虫转录组学研究也越来越受到重视。秀丽隐杆线虫[4]()基因组测序组织对不同发育阶段的模式生物进行基因组学研究,构建了基因组学图谱并详细注释出发育相关的功能基因。Fu等[5]对犬恶丝虫的转录组进行研究,组装出20 810个转录本,并发现有1 101个是犬恶丝虫特有的基因,为免疫抗原的发现提供帮助。Li等[6]对不同发育阶段的马来丝虫进行转录组测序,详细阐明了马来丝虫在不同发育阶段的转录表达模式,及差异表达基因的功能。Laing等[7-8]绘制出捻转血矛线虫()基因组及不同发育期转录本的草图,发掘出重要的疫苗和药物靶点。【本研究切入点】前人在多种寄生虫发育转录组学研究中取得了阶段性成果,但是对斯氏副柔线虫的研究还很匮乏,尤其是对其不同发育阶段转录组基因表达研究仍属于未知。【拟解决的关键问题】本研究通过对不同发育阶段斯氏副柔线虫进行转录组测序,试图揭示斯氏副柔线在传播媒介与终末宿主体内不同发育阶段虫体在代谢水平上的差异、参与的调控机制;发掘出吸血性线虫特有功能基因及生长发育相关基因,为斯氏副柔线虫病的相关基础理论研究、诊断方法及防治研究奠定基础。
1 材料与方法
试验于2014—2016年在内蒙古农业大学兽医学院完成。
1.1 3个不同发育阶段斯氏副柔线虫样本采集
选取斯氏副柔线虫虫卵、第三期幼虫(L3s)、雌虫3个阶段的虫体进行转录组测序。雌虫成虫样本为2014年和2015年11—12月间采集于内蒙古巴盟乌拉特后旗的双峰驼真胃中,在显微镜下鉴定出雌虫,分装标记后在液氮中保存。第三期幼虫为2014年和2015年的7—8月间采集于内蒙古巴盟乌拉特后旗驼群环境中的吸血角蝇体内,在显微镜下鉴定后保存于液氮中。虫卵的收集通过将成年雌虫置于37℃生理盐水中过夜产卵,次日收集、镜下鉴定和计数后液氮保存,用于RNA提取。
1.2 方法
1.2.1 RNA提取 参照Invitrogen 公司的Trizol Reagent说明书分别对不同发育阶段斯氏副柔线虫总RNA进行提取。使用RQ1酶消解RNA中的DNA,纯化后将样本稀释进行UV检测及1.5%普通琼脂糖凝胶电泳质检合格后备用。
1.2.2 建立cDNA文库及Illumina测序 利用oligodT-磁珠富集捕获带有polyA尾巴的mRNA。在高温盐离子作用下,mRNA被随机打断,修复并连接5′Adaptor,然后用带有3’Adaptor和随机六聚体的RT引物反转录合成cDNA。对反转录后的cDNA进行PCR扩增,扩增时引入barcode序列,最终选取片段大小为300—500 bp的PCR产物,利用Illumina HiSeq2000TM测序平台进行测序。
1.2.3 测序数据分析 将测序所得的原始数据进行质量评估和可信度分析,并去除测序过程中低质量的序列和不确定的序列(Q<20),将得到的Clean reads合并,利用Trinity软件做转录组重头组装,对样品组装得到的Unigenes做进一步序列拼接、去冗余处理和同源聚类,最终得到转录本。将3个样本Unigenes的表达量进行RPKM值归一化处理。
在差异表达基因(differentially expressed genes,DEG)的筛选中,使用edgeR软件进行两两样本间的差异表达分析。检测过程中,将差异倍数(fold change,FC)≥2 或≤0.5且值≤0.01作为筛选标准,并利用logCMP模型对两个样本进行标准化,这样可以避免不明确的值和不明确的少数趋向于零的logFC,使两样本之间的比较更加详细和全面。
1.2.4 差异表达基因注释分析 斯氏副柔线虫虫卵和L3s,L3s和雌虫相比较,将获得的差异表达基因分别进行GO功能注释,然后将其按细胞组分、分子功能和生物过程3个GO数据库做功能分类及富集分析;同时将差异表达基因进行 KEGG 富集分析,把差异显著的通路进行富集,找到不同发育阶段虫体内显著性差异
变化的生物学调控通路;将两组比较的差异表达基因与基因编码的蛋白分别进行比对,并注释。然后用能够被注释的差异基因所对应的蛋白GI号在DAVID平台进行功能聚类分析,获得可信度高的功能通路和更细致全面的功能聚类。
2 结果
2.1 斯氏副柔线虫Unigenes聚类后数据可靠性分析
本研究对斯氏副柔线虫虫卵、第三期幼虫(L3s)和成年雌虫3个发育阶段分别进行转录组测序,将得到的数据进行质量控制、拼接组装和聚类,总共获得99 481个Cluster Unigenes用于差异基因分析。使用RPKM法计算各个发育阶段Unigenes表达量。结果显示,聚类后的Unigenes在47%—76%之间都有reads的分布,虫卵、L3s、雌虫中RPKM值大于10的Unigenes占所有表达Unigenes的9%—22%左右,说明聚类后数据可靠性较高(表1)。
2.2 不同发育阶段斯氏副柔线虫的差异表达基因相关分析结果
2.2.1 不同发育阶段斯氏副柔线虫的差异表达基因筛选结果 选用edgeR软件分别对斯氏副柔线虫L3s和虫卵、雌虫和L3s比较的基因表达量做差异表达分析,以-value≤0.01及Fold Change≥2或≤0.5为标准筛选差异表达基因(图1)。图1中红色的点表示显著差异表达基因(DEGs),纵坐标为logFC表示某一个基因在两样本中表达量差异倍数的对数值,且绝对值越大,表明基因表达量变化的倍数越大;横坐标为logCPM表示两样本之间比较同一个基因时的总聚集点,且坐标值越大,表明筛选的差异表达基因越可靠。
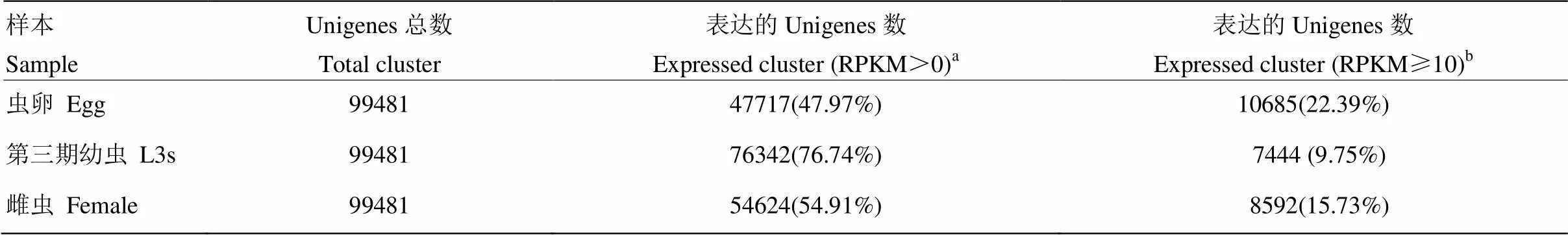
表1 每个样本表达的Unigene聚类
a表达的Unigenes数占总参考基因组Unigenes总数的比例;bRPKM值大于等于10的Unigenes数占RPKM值大于0的Unigenes的比例
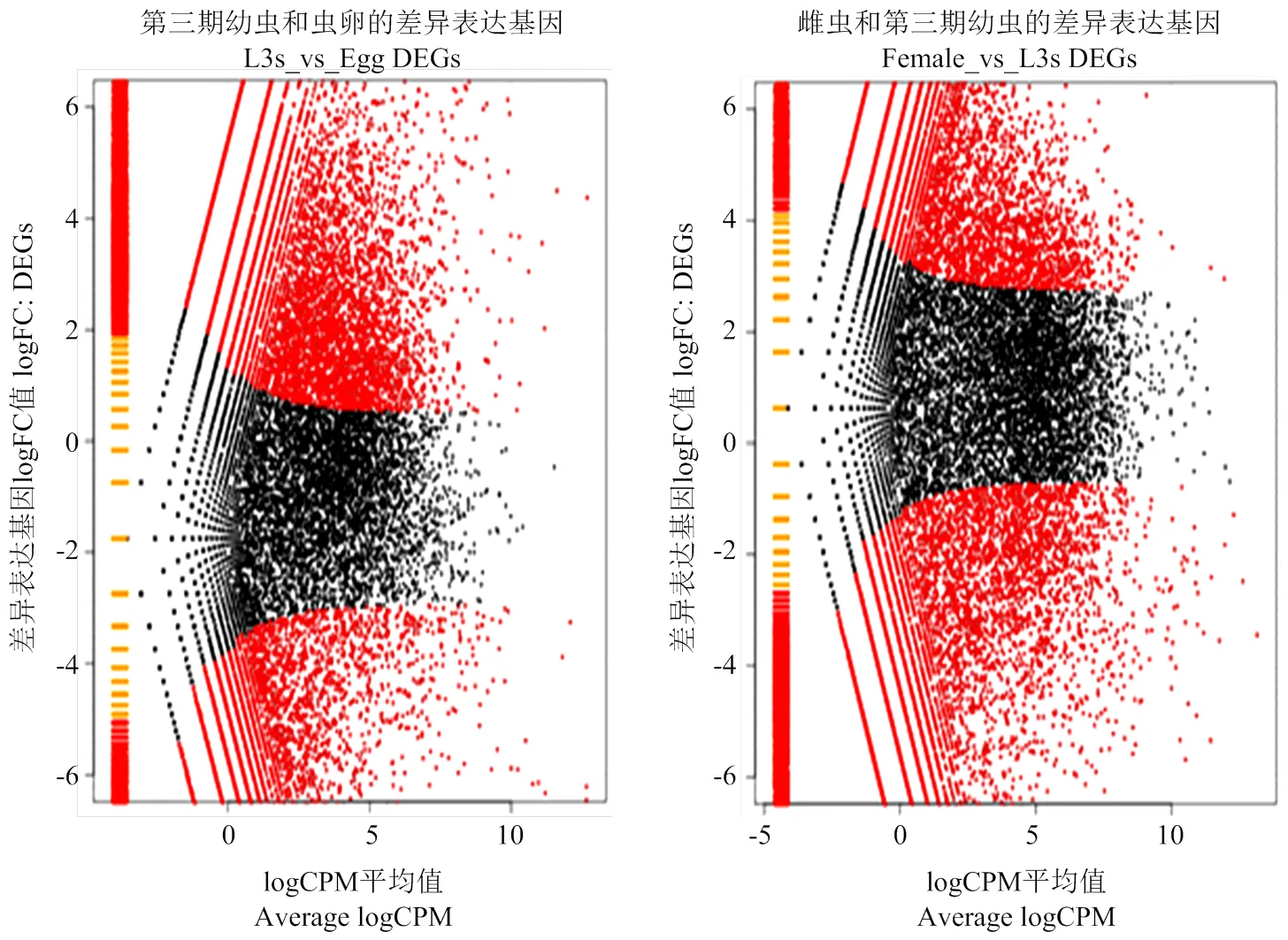
图1 edgeR方法鉴定虫卵和L3s、雌虫和L3s的差异表达基因
2.2.2 不同发育阶段斯氏副柔线虫差异基因表达结果 不同发育阶段斯氏副柔线虫成对比较后,将得到的差异表达基因进行统计,结果表明,L3s和虫卵相比,差异表达基因共有33 579个,表达上调的基因有20 477个,其中有7 561个上调基因注释出同源蛋白;表达下调的基因有13 102个,其中有2 645个下调基因注释出同源蛋白。雌虫和L3s比对时,差异表达基因共有32 199个,表达上调的基因有9 293个,其中3 874个上调基因注释出同源蛋白,表达下调的基因有22 906个,其中注释出6 384个下调基因注释出同源蛋白(图2)。
2.3 不同发育阶段斯氏副柔线虫差异表达基因Gene Ontology分析
2.3.1 不同发育阶段斯氏副柔线虫差异表达基因GO-生物过程富集分析 在L3s和虫卵的差异表达基因GO-生物过程富集分析中,有6 617个差异表达基因比对到BP数据库中,显著富集在蛋白质氨基酸磷酸化过程中的差异表达基因(DEG)有119个,占该生物过程基因数(EG)的51.74%,占比对到BP数据库所有差异表达基因数的1.80%;显著富集在翻译过程中的差异表达基因有130个,占该生物过程基因数的46.76%,占比对到BP数据库所有差异表达基因数的1.97%。雌虫和L3s比对中,有7 043个差异表达基因比对到BP数据库中,除了在翻译、肌肉组织发育等过程中显著富集,在染色体结构中有9个差异表达基因,占该生物学过程基因数的81.82%,占比对到BP数据库所有差异表达基因数的0.13%;有8个差异基因显著富集在性腺发育过程中,占该生物学过程基因数的80.00%,占比对到BP数据库所有差异表达基因数的0.11%(表2)。
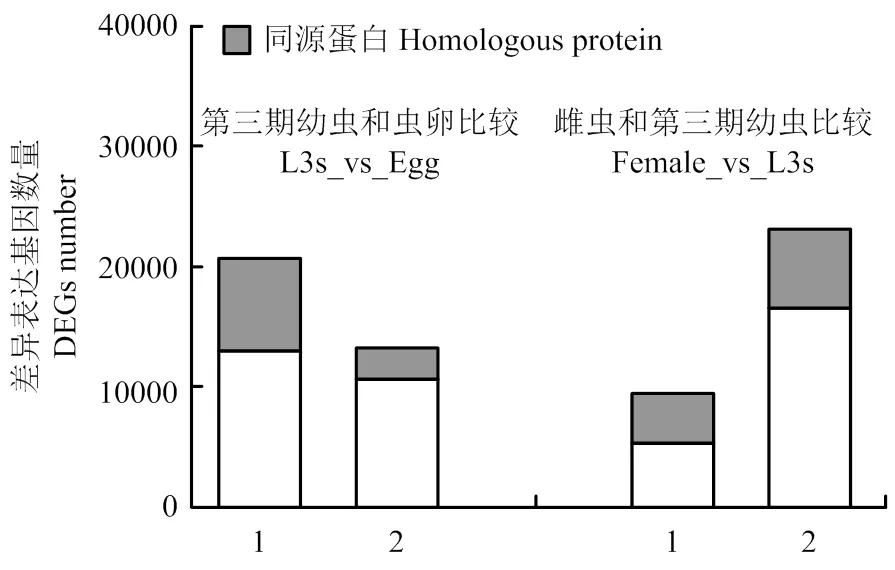
1:下调差异表达基因 Up-regulated DEGs;2:上调差异表达基因 Down-regulated DEGs
2.3.2 不同发育阶段差异表达基因GO-细胞组成富集分析 L3s和虫卵GO-细胞组分富集分析中,有3 891个差异表达基因比对到CC数据库,其中,有90个显著富集在细胞溶质中,占该细胞组分基因数的61.64%,占比对到CC数据库所有差异表达基因数的2.31%;显著富集在细胞核中的差异表达基因有215个,占该细胞组分基因数的47.46%,占比对到CC数据库所有差异表达基因数的5.53%;显著富集在线粒体中的差异表达基因有58个,占该细胞组分基因数的55.77%,占比对到 CC数据库所有差异表达基因数的1.49%。雌虫和L3s比对中,有3 686个差异表达基因比对到CC数据库。除了基本细胞组分的富集之外,有6个差异表达基因在细胞内无膜细胞器中显著富集,占该细胞组分基因数的100%,占比对到CC数据库所有差异表达基因数的0.16%。有20个差异表达基因显著富集在胶原蛋白中,占该细胞组分基因数的58.82%,占比对到CC数据库所有差异表达基因数的0.54%。
2.3.3 不同发育阶段差异表达基因GO-分子功能富集分析 L3s和虫卵GO-分子功能富集分析中,有8 755个差异表达基因比对到MF数据库。其中,有172个差异基因显著富集在蛋白结合功能中,占该分子功能基因数的47.78%,占比对到MF数据库所有差异表达基因数的1.97%;有77个显著富集在蛋白丝氨酸/苏氨酸激酶活性中,占该细胞组分基因数的52.38%,占比对到MF数据库所有差异表达基因数的0.88%;显著富集在亚铁血红素结合中的差异表达基因有38个,占该细胞组分基因数的48.10%,占比对到MF数据库所有差异表达基因数的0.43%。雌虫和L3s比对中,有10 177个差异表达基因比对到MF数据库,在蛋白结合、锌离子结合和亚铁血红素结合等分子功能中显著富集的同时,有18个差异表达基因在氢离子跨膜转运活性中显著富集,占该分子功能基因数的69.23%,占比对到MF数据库所有差异表达基因数的0.18%。有18个显著富集在表皮结构组成中,占该细胞组分基因数的69.23%,占比对到MF数据库所有差异表达基因数的0.18%(表2)。
2.4 不同发育阶段斯氏副柔线虫差异基因KEGG Pathway分析
将不同发育阶段斯氏副柔线虫的差异基因注释到KEGG数据库,结果显示,L3s和虫卵中比对到差异表达基因12 803个,其中6 521个差异表达基因有具体的定义,并显著富集到255条通路。其中氧化磷酸化通路显著富集221个差异表达基因,占该通路基因数的64.62%;MAPK信号通路显著富集100个差异表达基因,占该通路基因数的63.69%;Wnt信号通路显著富集90个差异表达基因,占该通路基因数的58.82%。雌虫和L3s中比对到13 153个差异表达基因,其中有6 528个差异表达基因有具体的定义,同时涉及251条通路。其中代谢通路显著富集有1 679个差异表达基因,占该通路基因数的51.61%,细胞循环通路显著基因有60个,占该通路的63.16%(表3)。
2.5 差异表达基因功能聚类
将斯氏副柔线虫L3s和虫卵、雌虫和L3s相比的差异表达基因利用DAVID平台进行功能聚类分析。结果显示,有关防御机制;己糖代谢、葡萄糖代谢、糖酵解/糖异生等功能聚类在L3s中富集程度明显高于虫卵和雌虫阶段,这可能与L3s期幼虫在感染哺乳动物宿主时,采取自身免疫保护和免疫逃避有关。雌虫和虫卵阶段在生长调控速率和生殖发育等相关功能聚类中富集性高表达,而L3s中富集不明显。雌虫在性别分化、生殖器发育等功能聚类中显著性高表达。虫卵、三期幼虫和雌虫在胚胎发育、胚后发育以及幼虫发育等功能聚类中都富集性高表达。在胚胎发育功能聚类中有242个相关功能基因在3个发育阶段都表达,而在虫卵、L3s和雌虫中分别有63个、571个和248个特异性表达的功能基因。在胚后发育功能聚类中有202个相关功能基因富集在3个发育阶段,详情见图3。
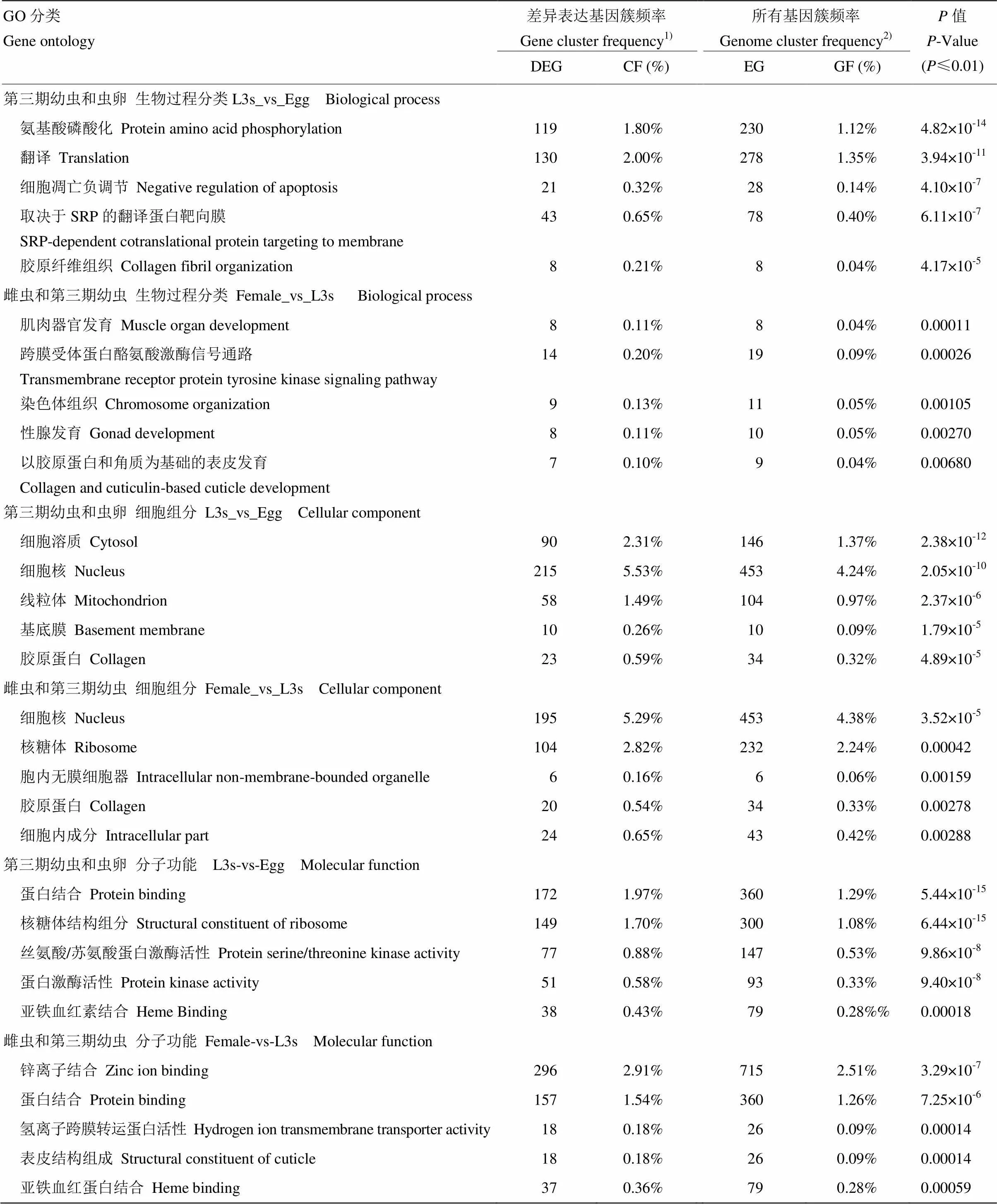
表2 L3s和Egg,雌虫和L3s差异表达基因GO-ontology富集分析
1)CF=DEG/TDEG×100%,CF:在该ontology中差异表达基因簇的频率;DEG:在该ontology中差异表达基因的数量;TDEG:比对到ontology中所有差异表达基因数。2)GF=EG/TEG×100%,GF:比对到该ontology中基因的频率;EG:比对到该ontology中所有基因数量;TEG:比对到ontology中所有基因数
1)CF=DEG/TDEG×100%, CF is cluster frequency of differentially expressed gene annotated to each ontology; DEG is the numbers of differentially expressed gene annotated to each ontology; TDEG is numbers of all differentially expressed genes annotated to GO ontology.2)GF=EG/TEG×100%,GF is the genome frequency of all genes annotated to the ontology; EG is the numbers of genes annotated to each ontology; TEG is the numbers of all genes annotated to GO ontology
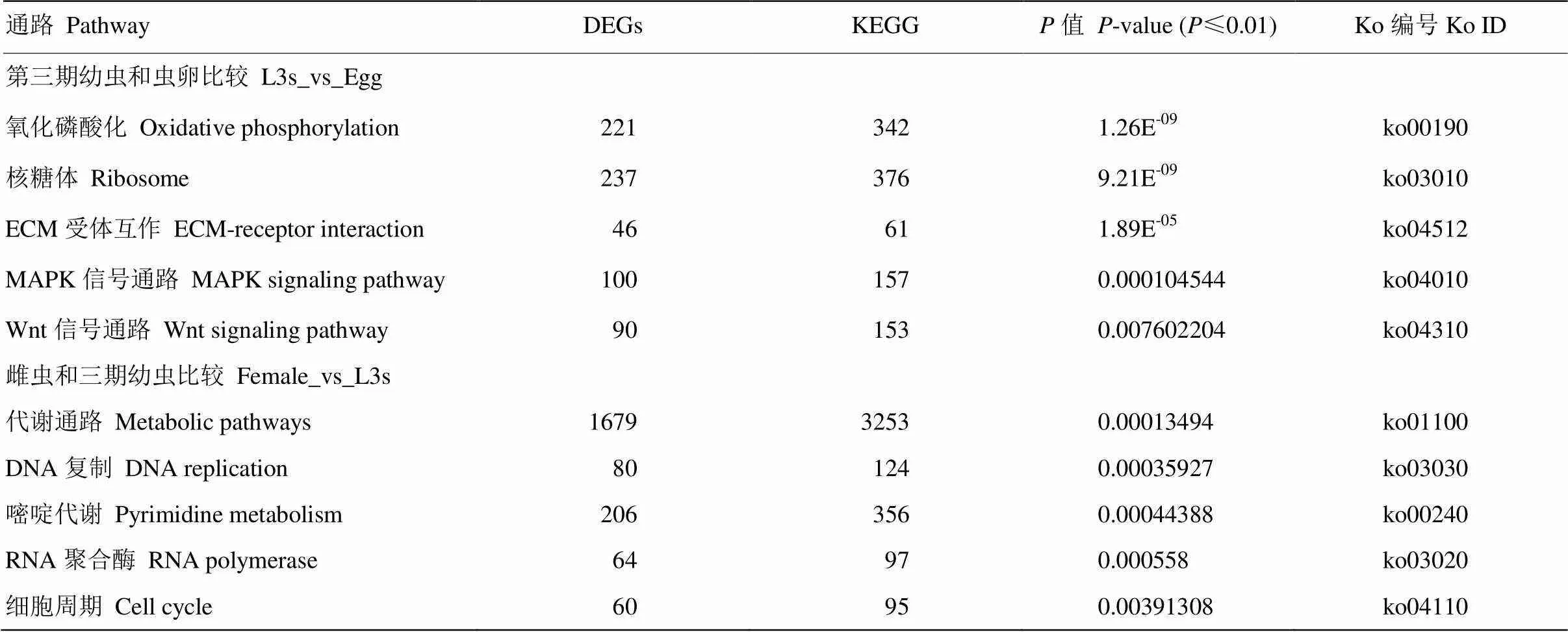
表3 L3s和虫卵,雌虫和L3s中差异表达基因显著性富集的通路
DEGs:在每个KEGG代谢通路中差异表达基因数量;KEGG:在KEGG数据库中涉及到该通路的基因数
DEGs: The numbers of Differentially expressed genes in each KEGG pathway; KEGG: Gene numbers in this pathway in KEGG database
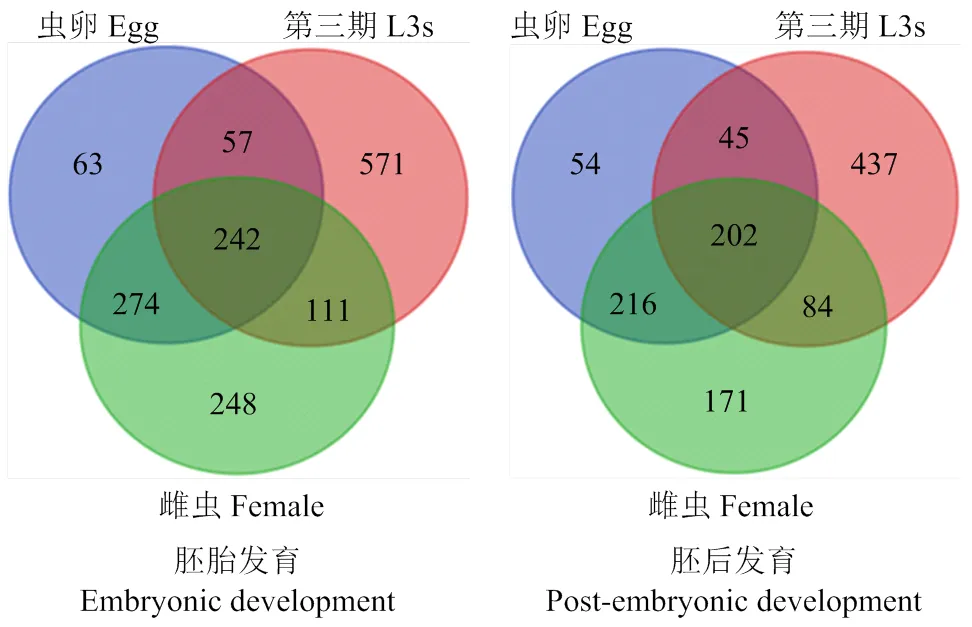
图3 虫卵、幼虫和雌虫在胚胎发育和胚后发育过程中相关功能基因的韦恩图分布
2.6 不同发育阶段斯氏副柔线虫生长发育相关基因分析
本文以影响生长发育相关的基因为参考,分析出不同发育阶段斯氏副柔线虫中生长发育相关的重要基因并对其进行富集分析。结果显示,在虫卵、L3s和雌虫3个发育阶段中都出现了不同表达程度的异时性相关基因: 核受体DAF-12、 LIN-12和BLMP-1基因在L3s中高表达;DRE-1/FBXO11和LIN-42基因在雌虫中高表达;LIN-29基因在虫卵中较高表达,在L3s和雌虫中几乎不表达;RHEB-1和LIN-28在虫卵和雌虫中高表达,在L3中表达量较低;LIN-14在3个发育阶段中都有表达,其中在L3s中表达量相对较高。
本研究分析了核激素受体(nuclear hormone receptor,NHRs)和锌金属蛋白酶(NAS)两类在线虫生长发育过程中重要的发育基因。NHRs是生物体内发育和代谢过程中重要的调节者,在斯氏副柔线虫3个发育阶段中共注释出48个核激素受体同源蛋白,其中有26个具有详细的功能分类,主要包括:NHR-49、NHR-48、NHR-40、NHR-1等,其中NHR-40和NHR-49在L3s期中较高表达,NHR-48在雌虫和L3s中相对高表达,NHR-1在虫卵和雌虫期中较高表达。锌金属蛋白酶,又可称作线虫虾红素(nematode astacin,NAS)对线虫表皮合成和表皮胶原蛋白酶裂解有重要作用。在斯氏副柔线虫3个发育阶段中注释出36个NAS,其中有26个有详细的功能分类,主要包括NAS-36、NAS-33和NAS-14等。其中NAS- 36在L3s期中相对高表达,在虫卵期和雌虫期未表达;NAS-15在虫卵和雌虫期的表达量高于L3s期,NAS-14在L3s期的表达量高于虫卵和雌虫期,具体详情见表4。
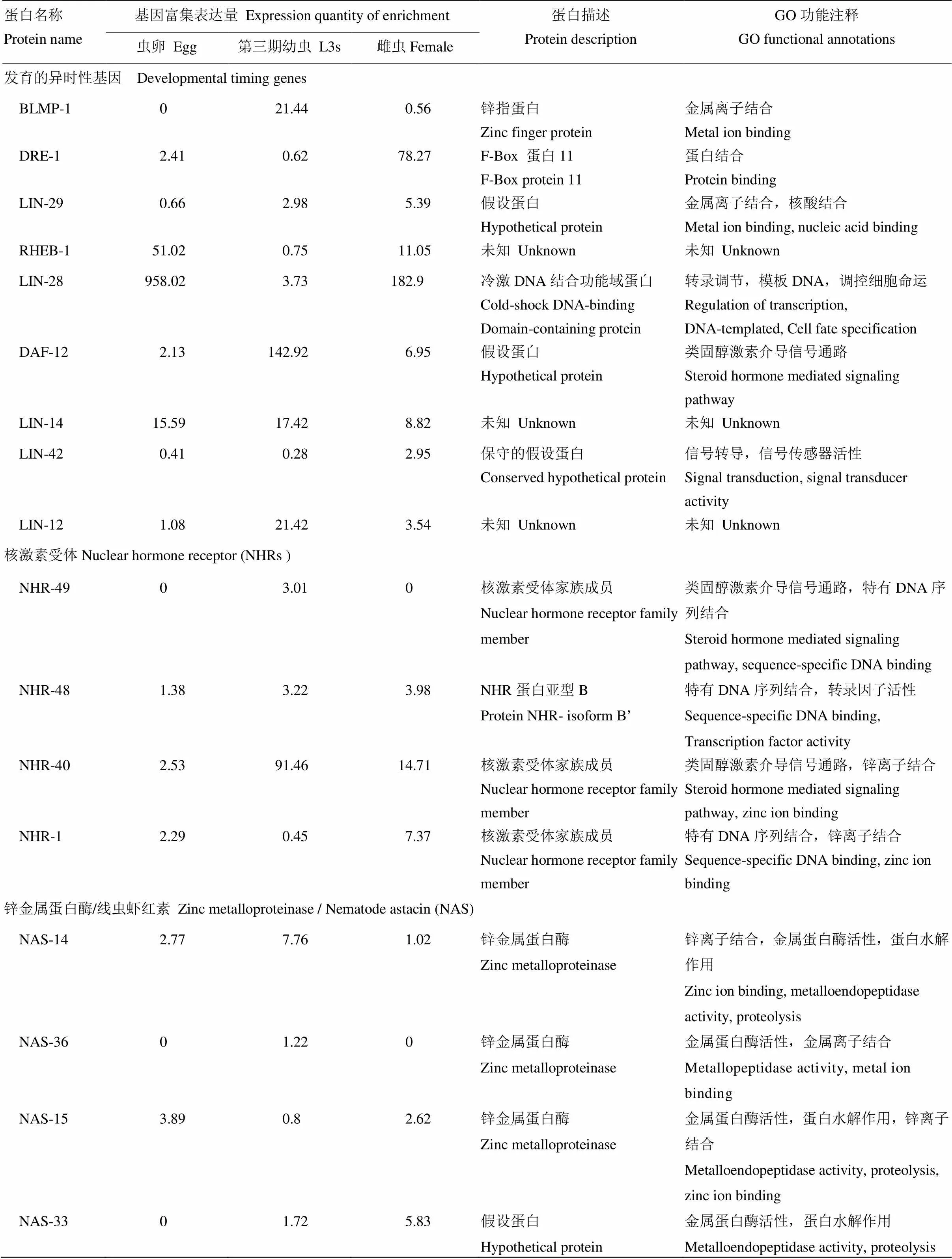
表4 虫卵期、幼虫期和雌虫期生长发育相关的重要基因
3 讨论
新一代高通量测序技术的不断发展,已彻底改变了转录组学的研究,使RNA-Seq无需预先设计探针即可对特定条件下任意生物生长发育阶段整体转录活动进行测序,并且探测各种条件下的基因表达情况,发现了许多未知的研究领域[9-10]。本研究针对虫卵、第三期幼虫和雌虫3个发育阶段的斯氏副柔线虫进行转录组测序分析,探索不同发育阶段中基因表达的差异、代谢通路的差异及生长发育过程中功能聚类的显著差异。样本采集的代表性对转录组数据的准确性和代表性起至关重要作用,所以该研究中样本采集是重要的质控过程。由于不同采集时间和样本自身特点等因素,导致不同时间采集的相同发育阶段虫体基因表达量可能存在一定的差异。因此,本研究为了使获取的转录本数据更加全面,将三个样本的采集次数均增加至两年中的9—10次;同时采集样本数量也增加到虫卵80万只左右,第三期幼虫5 000只左右,雌虫200只左右。通过将多次采集时间的大量样本进行混合上机测序,以保证转录本测序数据的全面性和代表性,提高数据的重复性和多样性。
3.1 差异表达基因的GO功能富集分析
不同发育阶段斯氏副柔线虫的差异表达基因在GO功能富集分析中显示,L3s和虫卵相比,显著富集到负调控细胞程序性死亡通路;而幼虫到雌虫富集明显减少,可能与其为了生长发育,加快代谢通路有关。雌虫和L3s相比,富集到肌肉器官发育、生殖腺发育、胶原蛋白和基于角质的表皮发育信号通路;而L3s和虫卵中富集明显减少,说明在幼虫到雌虫开始摄血与胶原蛋白、表皮发育、生殖腺发育相关的基因表达显著增加,这与SCHWARZ 等[8]对捻转血矛线虫的研究一致。在GO-细胞组分数据库中,L3s和虫卵相比,显著富集到细胞溶质、细胞核和胶原蛋白中;雌虫和L3s相比,显著富集到细胞核、核糖体及细胞内有膜细胞器中,而细胞核、核糖体与虫体生长发育有关。在GO-分子功能数据库中,L3s和虫卵相比,显著富集到蛋白质结合功能、钙离子结合功能和蛋白激酶活性中;雌虫和幼虫相比,显著富集到锌离子结合功能、氢离子跨膜转运蛋白活性及ATP结合功能,而蛋白激酶活性和氢离子跨膜转运蛋白活性与生长发育过程中能量消耗有关,表明在不同发育阶段虫体代谢耗能的主要方式不同,而ATP结合功能在幼虫到雌虫中显著富集,在虫卵到L3s中富集明显减少,可能与其发育阶段不同所需能量不同有关。从虫卵到L3s、L3s到雌虫的发育过程中亚铁血红素结合功能都显著富集,根据报道秀丽隐杆线虫中存在与脊椎动物(如鸽子、猪等)SCS-β亚基(丁二酰辅酶A连接酶)同源性较高的基因,GTP依赖型SCS在脊椎动物中参与三羧酸循环中的可逆反应,反向反应激活酮体亚铁血红素的合成[11-12],这与GO富集在亚铁血红素结合功能的结果一致。
3.2 差异表达基因的KEGG pathway富集分析
通过KEGG pathway对差异表达基因进行显著性富集分析,将差异显著的 pathway 进行富集,有助于找到不同发育阶段虫体中显著性差异变化的生物学调控通路。KEGG注释和聚类分析结果显示,L3s和虫卵相比富集到Wnt/MAPK信号通路,其通路中起重要作用的lit-1基因上调表达;根据报道Wnt/MAPK信号通路参与秀丽线虫侧线细胞的时序分化调控,如细胞命运特化和对称/不对称分裂[13-14],线虫中lit-1基因的缺失和增强会引起发育迟缓和发育过早的异时性缺陷[15-16],表明Wnt/MAPK信号通路富集在L3s发育阶段维持正常发育速度。雌虫和L3s相比主要富集在与生化代谢有关的代谢通路中,如嘌呤代谢、嘧啶代谢;与遗传信息有关的DNA复制、RNA聚合酶和细胞周期通路中;与糖异生信号有关通路中。已报道捻转血矛线虫L1到L3时期以及秀丽隐杆线虫Dauer时期糖异生作用显著增强[17],而斯氏副柔线虫在雌虫和幼虫相比中有显著富集,可能由于物种差异导致代谢通路在时空性上存在差异。
3.3 差异表达基因功能聚类分析
本研究利用DAVID平台进行功能聚类分析,DAVID的功能聚类数据库整合了Gene Ontology、Interpro、KEGG等基因功能数据库,获得的功能通路可信度高并且全面细致。同时将繁多的聚类获得的Clusters 进行翻译和归类,以便进行不同样品之间的比较。功能聚类结果分析显示,雌虫在生殖发育、性别分化、生殖器发育、雌雄同体的生殖器发育中相关基因高表达,而在L3s中低表达,其中雌雄同体生殖器发育相关基因对于线虫的性别分化发挥重要的功能,与性别有关的基因小窝蛋白(CAV-1)在雌虫中上调表达,这与秀丽隐杆线虫发现CAV-1在胚胎和生殖细胞中高表达[18]以及CAV-1在旋毛虫中高表达的研究一致[19]。在L3s和虫卵中,发现ACT-4基因在L3s和卵中富集性高表达,而在雌虫和L3s中富集性低表达,根据报道在秀丽隐杆线虫中,ACT-4基因在低氧胁迫条件下表达量上升,与LEV-11、MLC-1一起发挥细胞骨架的功能[20],所以在卵和L3s中可能由于低氧环境ACT-4基因表达量上升。
3.4 不同发育阶段斯氏副柔线虫生长发育相关基因分析
以模式生物秀丽隐杆线虫为参考,发现斯氏副柔线三个发育阶段中表达出不同程度的异时性相关基因。经分析发现,LIN-14基因存在于斯氏副柔线虫3个发育阶段中,这可能与LIN-14在胚胎发育后期分开调控发育时期特异性活动有关,且LIN-14过早或迟缓发育的突变可使雌虫期外阴发育迟缓,影响产卵系统[21]。LIN-14抑制LIN-12活性,LIN-14通过LIN-12对外阴前体细胞(VPCs)的时间控制来作用于VPCs的空间发育模式,两者共同调控外阴前体细胞的命运[22]。RHEB-1基因在斯氏副柔线虫虫卵和雌虫期表达量较高,这可能与其调控线虫寿命长度的功能有关[23];LIN-42基因在斯氏副柔线虫雌虫期相对高表达,这可能与LIN-42调控线虫性腺发育,外阴及性肌母细胞的发育时序功能有关[24];BLMP-1和 DRE-1/FBOX11在斯氏副柔线虫3个发育阶段的表达量呈负相关,这与2014年Horn等发现通过锌指蛋白BLMP-1 与DRE-1/FBOX11结合而被降解来控制秀丽隐杆线虫的时空发育、dauer形成、蜕皮、性腺的成熟和寿命的研究一致[25],说明两个蛋白之间呈负相关相互调控,在后生生物成熟过程中发挥作用。2015年WANG等[26]对线虫类固醇激素受体DAF-12基因研究发现,在适宜的环境条件下,DAF-12被DA(dafachronic acids)激活,DAF-12的活化作用能够协调动能的储存,从而使幼虫生长繁殖;在斯氏副柔线虫L3s期中DAF-12基因高表达,由于L3s在中间宿主体内适应寄生环境后,L3s中DAF-12被活化,存储动能使幼虫生长发育。HUANG等[27]研究发现顶端细胞(DTCs)对性腺的形成有重要的作用,BLMP-1负调控DTCs背向弯曲,而DAF-12,DRE-1,LIN-29功能冗余,正调控背向弯曲。DAF-12和LIN-29抑制BLMP-1转录,而DRE-1结合到BLMP-1促进其降解,使BLMP-1负调控获得正确调控时间。在器官形成期间,不同的异时性基因将时间的和空间的信号整合成一个回路共同协调生物体整体发育。这些异时性相关基因形成了复杂的调控网络,调控线虫整个发育过程的时间性和空间性,保证各种器官的准确定位和适时发育。核激素受体(NHRs)是调控应对发育、环境及营养状况信号的基因表达蛋白,NHR对很多发育过程有重要作用,其中NHR-49 具有调节和控制脂肪代谢、维持脂肪酸饱和的正常平衡功能[28];在肠道中抑制溶酶体脂质的累积,与PKG信号通路共同调控短期禁食诱导溶酶体脂质的积累[29];并且NHR-49调控胚胎发育、幼虫发育及成虫寿命。NHR-49在斯氏副柔线虫L3s期中较高表达,可能与其调控幼虫发育有关。NHR-48参与转录本调控并具有序列特异的DNA结合活性和转录因子活性等功能。根据BROZOVA等[30]研究发现NHR-40主要调控晚期胚胎和早期幼虫的延长和形态发生,且NHR-40功能表型缺失会导致不规律的体壁肌肉细胞发育,损害运动和神经肌肉协调。在线虫的不同幼虫期发育中锌金属蛋白酶对矫正发育、表皮适当蜕皮起着至关重要的作用[31]。NAS-36是控制蜕皮的关键基因,其突变后可扰乱蜕皮过程,使线虫无法进入正常的发育阶段,并且NAS-36还可以通过加工特定的细胞外基质蛋白调控新的表皮;在寄生性线虫期间NAS-36也是关键的靶基因,可用来控制杀虫效应。在斯氏副柔线虫中NAS-36主要在L3s期高表达,根据Stepek等[32-33]研究发现,NAS-36基因无论是在自生性线虫还是在寄生性线虫中基因功能都较保守,所以NAS-36在L3s期中也是控制蜕皮的关键基因。
4 结论
本研究绘制出斯氏副柔线虫在虫卵、第三期幼虫和雌虫3个发育阶段的差异基因表达谱;分析了斯氏副柔线虫在传播媒介和终末宿主体内不同发育阶段代谢水平上的差异;探索了不同发育阶段虫体之间重要的功能聚类,并注释出生长发育相关的功能基因,为斯氏副柔线虫相关理论研究、药物靶点发掘、免疫学诊断和防治研究奠定了基础。
[1] 宋铭忻, 张龙现. 兽医寄生虫学. 北京: 科学出版社, 2009: 183-184.
SONG M X, ZHANG L X.Beijing: Science Press, 2009: 183-184. (in Chinese)
[2] 黄兵, 沈杰. 中国畜禽寄生虫形态分类图谱. 北京: 中国农业科学技术出版社, 2006: 462-463.
HUANG B, SHEN J.Beijing: China Agriculture Press, 2006: 462-463. (in Chinese)
[3] 赵治国. 我国骆驼斯氏副柔线虫病传播媒介的研究[D]. 内蒙古农业大学, 2010.
Zhao Z G. Study on the vector of camel parabronemosis in China[D]. Inner Mongolia: Inner Mongolia Agricultural University, 2010. (in Chinese)
[4] elegans Sequencing ConsortiumC. Genome sequence of the nematode: a platform for investigating biology., 1998, 282: 2012–2018.
[5] Fu Y, Lan J, Zhang Z, Hou R, Wu X, Yang D, Zhang R, Zheng W, Nie H, Xie Y, Yan N, Yang Z, Wang C, Luo L, Liu L, Gu X, Wang S, Peng X, Yang G. Novel insights into the transcriptome of, 2012, 7(7): e41639.
[6] Li B W, Wang Z Y, Rush C A, Mitreva M, Weil J G. Transcription profiling reveals stage-and function-dependent expression patterns in the filarial nematode, 2012, 13: 184.
[7] Laing R, Kikuchi T, Martinelli A, Tsai IJ, Beech R N, Redman E, Holroyd N, Bartley D J, Beasley H, Britton C, Curran D, Devaney E, Gilabert A, Hunt M, Jackson F, Johnston SL, Kryukov I, Li K, Morrison A A, Reid A J, Sargison N, Saunders G I, Wasmuth J D, Wolstenholme A, Berriman M, Gilleard J S, Cotton J A. The genome and transcriptome of, a key model parasite for drug and vaccine discovery., 2013, 14: R88.
[8] Schwarz E M, Korhonen P K, Campbell B E, Young N D, Jex A R, Jabbar A, Hall R S, Mondal A, Howe A C, Pell J, Hofmann A, Boag P R, Zhu X Q, Gregory T, Loukas A, Williams B A, Antoshechkin I, Brown C, Sternberg P W, Gasser R B. The genome and developmental transcriptome of the strongylid nematode., 2013, 14(8): R89.
[9] 刘娇, 张建珍, 李大琪, 张婷婷, 马恩波, 张建琴. 中华稻蝗羧酸酯酶家族基因生物信息学及组织表达特异性分析. 中国农业科学, 2015, 48(21): 4272-4284.
LIU J, ZHANG J Z, LI D Q, ZHANG T T, MA E B, ZHANG J Q. Bioinformatics and tissue-specific expression analysis of carboxylesterase genes from., 2015, 48(21): 4272-4284. (in Chinese)
[10] 陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 张曌楠, 黄枳腱, 张璐, 王鸿权, 解彦玲, 童新宇. 中华蜜蜂幼虫肠道响应球囊菌早期胁迫的转录组学. 中国农业科学, 2017, 50(13): 2614-2623.
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENG Y Z, XU X J, ZHANG Z N, HUANG Z J, ZHANG L, WANG H Q, XIE Y L, TONG X Y. Transcriptome oflarval gut under the stress of., 2017, 50(13): 2614-2623. (in Chinese)
[11] Johnson J D, Mehus J G, Tews K, Milavetz B I, Lambeth D O. Genetic evidence for the expression of ATP- and GTP-specific succinyl-CoA synthetases in multicellular eucaryotes.1998, 273(42): 27580-27586.
[12] Przybyla Z B, Dennis R A, Zakharkin S O, McCammon M T. Genes of succinyl-CoA ligase from., 1998, 258(2): 736-743.
[13] Delawary M, Nakazawa T, Tezuka T, Sawa M, Iino Y, Takenawa T, Yamamoto T. Molecular characterization of a novel RhoGAP, RRC-1 of the nematode., 2007, 357(2): 377-382.
[14] Yang X D, Karhadkar T R, Medina J, Robertson S M, Lin R. β-Catenin-related protein WRM-1 is a multifunctional regulatory subunit of the LIT-1 MAPK complex., 2015, 112(2): E137-146.
[15] Chuang M H, Chiou S H, Huang C H, Yang W B, Wong C H. The lifespan-promoting effect of acetic acid and Reishi polysaccharide., 2009, 17(22): 7831-7840.
[16] Jackson B M, Abete L P, Krause M W, Eisenmann D M. Use of an activated beta-catenin to identify Wnt pathway target genes in, including a subset of collagen genes expressed in late larval development., 2014, 4(4): 733-747.
[17] John W, Stuart K K. Global analysis of dauer gene expression in., 2003, 130: 1621-1634.
[18] Parker S, Baylis H A. Overexpression of caveolins ininduces changes in egg-laying and fecundity., 2009, 2(5): 382-384.
[19] Romel H B, Ricardo R N, Saé M H, Karen N C, Lenin P, Ana G S, Jorge M M. Sex steroids effects on the molting process of the helminth human parasite
[20] Li H, Ren C, Shi J, Hang X, Zhang F, Gao Y, Wu Y, Xu L, Chen C, Zhang C. A proteomic view ofcaused by short-term hypoxic stress., 2010, 8: 49.
[21] Johnson R W, Liu L Y, Hanna R W, Chamberlin H M. Theheterochronic gene lin-14 coordinates temporal progression and maturation in the egg-laying system., 2009, 238(2): 394-404.
[22] Li J, Greenwald I. LIN-14 inhibition of LIN-12 contributes to precision and timing ofvulval fate patterning., 2010, 20(20): 1875-1879.
[23] Honjoh S, Yamamoto T, Uno M, Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in., 2009, 457(7230): 726-730.
[24] Tennessen J M, Gardner H F, Volk M L, Rouqvie A E. Novel heterochronic functions of theperiod- related protein LIN-42., 2006, 289(1): 30-43.
[25] Horn M, Geisen C, Cermak L, Becker B, Nakamura S, Klein C, Pagano M, Antebi A. DRE-1/FBXO11-dependent degradation of BLMP-1/BLIMP-1 Governsdevelopmental timing and maturation., 2014, 28(6): 697-710.
[26] Wang Z, Jonathan S, You Y J, Ranjit N, Tang H, Xie Y, Lok J B, Mangelsdorf D J, Kliewer S A. The nuclear receptor DAF-12 regulates nutrient metabolism and reproductive growth in nematodes., 2015, 11(3): e1005027.
[27] Huang T F, Cho C Y, Cheng Y T, Huang J W, Wu Y Z, Yeh A Y, Nishiwaki K, Chang S C, Wu Y C. BLMP-1/Blimp-1 Regulates the spatiotemporal cell migration pattern in, 2014, 10(6): e1004428.
[28] Van G M R, Hadjivassiliou H, Jolly A, Yamamoto K R. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in.2005, 3(2): e53.
[29] Huang W M, Li Z Y, Xu Y J, Wang W, Zhou M G, Zhang P, Liu P S, Xu T, Wu Z X. PKG and NHR-49 signalling co-ordinately regulate short-term fasting-induced lysosomal lipid accumulation in., 2014, 461(3): 509-520.
[30] Brozova Z, Simeckova K, Kostrouch Z, Rall J E, Kostrouchova M. NHR-40, asupplementary nuclear receptor, regulates embryonic and early larval development., 2006, 123(9): 689-701.
[31] Suzuki M, Sagoh N, Iwasaki H, Inoue H, Takahashi K. Metalloproteases with EGF, CUB, and thrombospondin-1 domains function in molting of., 2004, 385(6): 565-568.
[32] Stepek G, McCormack G, Birnie A J, Page A P. The astacin metalloprotease moulting enzyme NAS-36 is required for normal cuticle ecdysis in free-living and parasitic nematode., 2011, 138(2): 237-248.
[33] Sharma O P, Agrawal S, Kumar M S. Physicochemical properties of the modeled structure of astacin metalloprotease moulting enzyme NAS-36 and mapping the druggable allosteric space of,andvia molecular dynamics simulation., 2013, 5(4): 312-323.
(责任编辑 林鉴非)
The Comparative Transcriptome Analysis ofat Different Developmental Stages
WANG WenLong1, FENG ChenChen1, HONG Mei1, YUE JianWei1, Huhebateer1, LIU ChunXia2
(1College of Veterinary Medicine, Inner Mongolia Agricultural University/Key Laboratory of Clinical Diagnosis and Treatment Technology in Animal Disease, Ministry of Agriculture, Hohhot 010018 ;2College of Life Sciences, Inner Mongolia Agricultural University, Hohhot 010018)
The objective of this study was to identify the differentially expressed genes(DEG) and describe biological characteristics involved in functional classifications and metabolic pathways at different developmental stages ofinfecting camel, which is necessary to better understand functional genes involved in growth and development and enrich the transcriptome data of parasitical nematodes.Eggs, the third-stage larvae(L3s) and females ofwere sequenced by Illumina HiSeq2000TMsequencing platform and constructed their cDNA libraries after quality filtering. De novo assembling and assembly efficiency assessment were carried out using Trinity, a short-read assembly program. Then all the effective sequential data obtained wereassigned to the relevant databases to perform functional annotation and bioinformatic analysis.The results showed that 47 717, 76 342 and 54 624 unigenes were obtained respectively in eggs,the third-stage larvae and female stages. 33 579 differentially expressed genes(DEGs) were identified by comparing the unigenes obtained from eggs and L3s stages, of which 20 477 were up-regulated and 13 102 were down-regulated. There were 32 199 differentially expressed genes between L3s and female stages, of these genes, 9 293 were up-regulated genes and 22 906 were down-regulated genes. The differentially expressed genes of two pairwise comparisons were respectively enriched in Gene Ontology. 6 617, 3 891 and 8 755 differentially expressed genes comparing eggs and L3s stages were annotated into database of biological process, cellular component and molecular function respectively, while the number by comparing L3s and female stages were 7 043, 3 686 and 10 177 respectively. In KEGG pathways identification, 6 521 differentially expressed genes comparing eggs and L3s stages were assigned to 251 KEGG pathways, and clustered significantly in MARK, Wnt signaling pathways and oxidative phosphorylation. In comparison of L3s and female stages, 6 528 differentially expressed genes were enriched in metabolic pathways, DNA replication and cell cycle. Functional cluster analysis indicated that the regulation related differential genes of growth rate and the reproductive and genital development were highly expressed in eggs and females stages, then the genes of the defense and carbohydrate metabolism were enriched in exclusive L3s stage. Differential genes of embryonic and post-embryonic development were highly expressed in all three stages, and 196 function genes from embryonic development and 166 function genes from post-embryonic development were co-expressed in all three stages, implied that these genes played a crucial role in (post-)embryonic development. Moreover, we identified 9 heterochronic genes, such as LIN-28, LIN-14 and RHEB-1, 48 nuclear hormone receptors (NHRs) genes, including NHR-49, NHR-48, NHR-40 and NHR-1, and 36 zinc metalloproteinase(NAS) genes, including NAS-36, NAS-33 and NAS-14. Then the analysis of enrichment capacity in three stages showed these genes were necessary to regulate the different development stages ofThe transcriptomic research ofat three developmental stages using RNA-seq revealed biological characteristics of differentially expressed genes involved in development- related GO functional classification, KEGG pathway and functional cluster and identified many kinds of heterochronic genes and developmental genes, which provides a foundation and reference for further investigation of the whole genome sequence analysis, interaction betweenand host, pathogenic mechanism and immune evasion.
; transcriptome; differentially expressed genes; development-related genes
2016-06-20;
2017-10-30
国家自然科学基金(31260603)
联系方式:王文龙,E-mail:wwl.imau@163.com。冯陈晨,E-mail:410580576@qq.com。王文龙和冯陈晨为同等贡献作者。通信作者呼和巴特尔,Tel:0471-4303726;E-mail:hhbte@163.com。通信作者刘春霞,Tel:0471-4309240;E-mail:lcx.imau@163.com
