高致猝死基因TNNT2基因Phe87Tyr突变致肥厚型心肌病1例
2017-12-15袁帅邢晓博王芳赵雯娜刘福颂宋雷
袁帅,邢晓博,王芳,赵雯娜,刘福颂,宋雷
高致猝死基因TNNT2基因Phe87Tyr突变致肥厚型心肌病1例
袁帅1,邢晓博1,王芳1,赵雯娜1,刘福颂1,宋雷2
肥厚型心肌病(HCM)是一种常染色体遗传性疾病,其后代患病和携带突变基因的风险约50%[1],以左心室壁增厚为主要表现,且通常无心脏负荷增加的表现,是年轻人发生心源性猝死(SCD)的主要原因之一,患病率约1:500。HCM患者发生房颤等心律失常和心力衰竭的风险常伴随终生[2]。HCM是首个明确致病基因的单基因心脏病[3],主要由编码肌小节结构蛋白的基因突变引起,至少有20种致病基因的400余种突变参与致病。国外研究结果[4-6]显示:Troponin T基因(TNNT2)为常见的致病基因之一,约占10%~20%。目前已发现30余种TNNT2基因突变与HCM发生有关,集中分布在8、9、10、11、14、15、16号外显子上。国内[7,8]有关TNNT2基因突变的报道较少,故本文就我们救治的1例TNNT2基因Phe87Tyr突变的男性患者进行报道。
1 病例
患者男性,56岁,主因“反复胸闷、憋气2年,加重1周”于2016年5月就诊于青岛市第三人民医院心内科门诊。患者于2年前反复出现胸闷、憋气,活动后加重,无胸痛及心悸不适,无夜间阵发性呼吸困难,无黑曚、晕厥发作,近1周上述症状加重,活动耐量稍有下降,夜间尚可平卧入睡。心电图示:窦性心律,左心室肥大。查血常规、凝血功能、肝肾功能、心肌酶谱、NT-proBNP均在正常范围。心脏超声示:室间隔中段增厚舒张期厚度15 mm,左室后壁中段舒张期厚度9.2 mm,考虑非梗阻性HCM。左房增大,左室舒张功能减退,二尖瓣反流(少量),如图1所示。24 h动态心电图示:窦性心律,偶发房性早搏。患者早在24岁行心脏超声时诊断为肥厚型心肌病。经家系调查发现,其家系有猝死史,其母于46岁猝死;其妹19岁猝死;其子14岁猝死,三者猝死前均有不同程度的心力衰竭症状,其父、其姐健在;患者因“心力衰竭”就诊我院心内科,予口服“倍他乐克、代文”改善心功能治疗后病情平稳,定期门诊随访。因发现该家系高猝死风险,我们对其进行基因验证检测,通过对肥厚型心肌病相关的30个致病基因的编码区及侧翼区采用2×150 Pair-End的模式进行第二代测序(Next generation sequencing,NGS),测序仪器为MiSeq System(Illumina公司),Sanger法对突变基因的相关区间进行测序验证。突变位点的确定运用http://www.ncbi.nlm.nih.gov/blast/bl2seq程序来分析测序结果,明确突变位点。检测结果发现该受检者携带TNNT2基因p.F87Y杂合错义变异(TNNT2:p.F87Y het),MYBPC3基因p.M1199I杂合错义变异(MYBPC3c.G3597T),如图2所示。
2 讨论
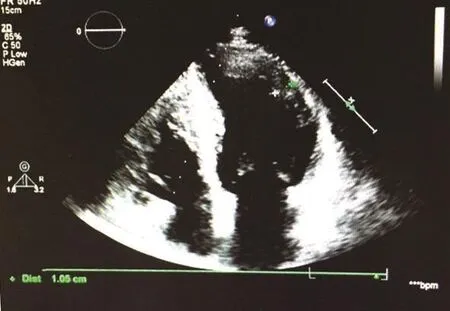
图1 患者超声心电图
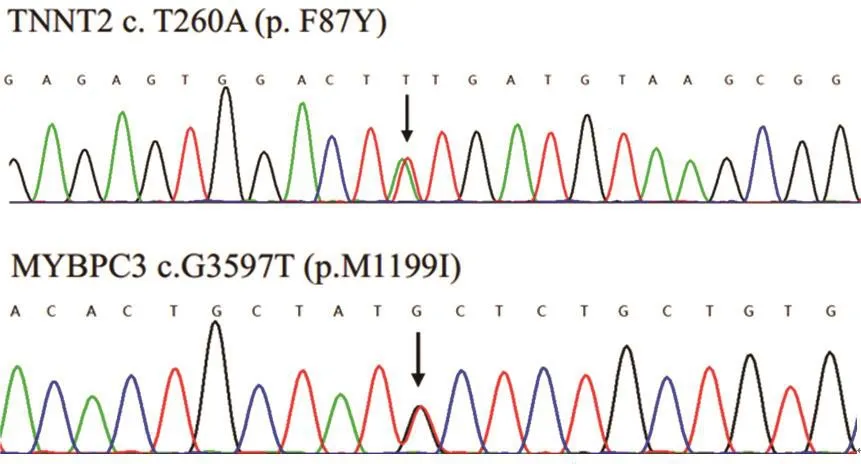
图2 Sanger测序图(箭头为突变位置)
肥厚型心肌病以心肌进行性肥厚、心室腔进行性缩小为特征,以左心室血液充盈受阻、舒张期顺应性下降为基本病理特点。其临床表现多样,可从无症状性左心室肥厚,到心律失常(包括心房纤颤、恶性室性心动过速)、难治性心力衰竭,部分患者首发症状即为猝死[9,10],是35岁以下人群猝死的主要原因。该病呈常染色体显性遗传,杂合致病变异携带者其子女遗传该变异的可能性为50%。肌钙蛋白T基因[11]位于染色体1q32,由16个外显子组成,编码蛋白是肌钙蛋白T,此蛋白与肌钙蛋白C、肌钙蛋白I组成肌钙蛋白复合物。TNNT2基因编码肌钙蛋白复合物的原肌球蛋白结合亚单位,位于横纹肌细肌丝,通过对细胞内钙离子浓度改变的反应来调节肌肉收缩[12]。
欧洲学者[13]在一有猝死发生的HCM家系中检测到p.F87L het,在这个家系中,共18例患者携带该变异基因,11例死于肥厚型心肌病(7例猝死、3例心力衰竭、1例卒中),在7例猝死患者中4例猝死年龄集中在14~16岁。18例患者中10例做过心超检查,7例心脏轻度肥大,3例心脏壁厚度>20 mm。11例中有50%在40~50岁时出现了呼吸困难。而家系中未携带该变异的成员均身体健康。国外学者[14,15]发现约5%~15%的HCM患者的致病基因为TNNT2,这类患者虽仅有轻度左室肥厚,但猝死发生率却较高。
本文患者携带的变异TNNT2:p.F87Y het导致第87位氨基酸由疏水性的苯丙氨酸变为亲水性的酪氨酸(所编码氨基酸的极性发生改变),国内外鲜有相关文献报道。已证实TNNT2附近位点的突变(V85L,D86A,I90M,R92W)被报道与HCM的发病有关,提示该变异所在基因区域的功能重要性。TNNT2:p.F87Y het千人基因组、ESP6500和ExAC人群频率数据库中均未见人群频率报道,查询本地数据库发现对照人群均未携带该变异,可明确其为罕见变异;该氨基酸同一位点的变异TNNT2:p.F87L het(由苯丙氨酸变为亮氨酸,极性未发生改变)被Clinvar数据库收录,判定为HCM的致病变异。
本例患者家族中其母,妹妹及其子,均有HCM的临床表现,且发生猝死,与文献报道TNNT2基因突变致病左室肥厚轻、猝死发生率高相一致,再结合该基因是HCM的重要致病基因以及该变异对蛋白产物的影响,应警惕该变异是一个HCM的高度可疑致病变异的可能。为进一步明确该变异是否为HCM的致病变异,可将该变异进行家系筛查,看家系中是否存在疾病与变异的共分离现象,但此患者其余亲属均拒绝。此患者同时携带了HCM临床意义尚不清楚的MYBPC3:p.M1199I het变异,应警惕两个变异叠加导致HCM加重的可能性。
故针对本例患者及其家系中的其他人群,应定期随诊心脏超声及动态心电图检查,注意心功能的变化,关注是否有恶性心律失常的存在,预防心源性猝死的发生。
[1]Seidman CE,Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy:a personal history[J]. Circ Res,2011,108(6):743-50.
[2]Gersh BJ,Maron BJ,Bonow RO,et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy:executive summary:a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines[J].Circulation,2011,124(24):2761-96.
[3]Coats CJ,Elliott PM. Genetic biomarkers in hypertrophic cardiomyopathy[J]. Biomark Med,2013,7(4):505-16.
[4]Mattos BP,Scolari FL,Torres MA,et al. Prevalence and Phenotypic Expression of Mutations in the MYH7,MYBPC3 and TNNT2 Genes in Families with Hypertrophic Cardiomyopathy in the South of Brazil:A Cross-Sectional Study[J]. Arq Bras Cardiol,2016,107(3):257-65.
[5]Fujita E,Nakanishi T,Nishizawa T,et al. Mutations in the cardiac troponin T gene show various prognoses in Japanese patients with hypertrophic cardiomyopathy[J].Heart Vessels,2013,28(6):785-94.
[6]Rubattu S,Bozzao C,Pennacchini E,et al. A Next-Generation Sequencing Approach to Identify Gene Mutations in Early-and Late-Onset Hypertrophic Cardiomyopathy Patients of an Italian Cohort[J].Int J Mol Sci,2016,17(8):1239.
[7]Jie L,Wenling L,Dayi H,et al. Mutation and clinical relevance in a large cohort of unrelated Chinese patients with hypertrophiccardiomy opathy[J]. Zhonghua Xin Xue Guan Bing Za Zhi,2015,43(8):682-9.
[8]Zhao Y,Cao H,Song Y,et al. Identification of novel mutations including a double mutation in patients with inheritedcardiomyopathy by a targeted sequencing approach using the Ion Torrent PGM system[J]. Int J Mol Med,2016,37(6):1511-20.
[9]Vatutin N,Taradin GG,Maron MS,et al. Sudden Cardiac Death in Patients With Hypertrophic Cardiomyopathy[J]. Kardiologiia,2016,56(1):56-65.
[10]Guo X,Fan C,Wang Y,et al. Genetic anticipation in a special form of hypertrophic cardiomyopathy with sudden cardiac deathin a family with 74 members across 5 generations[J]. Medicine,2017,96(11):e6249.
[11]李敏,程宽,王齐兵,等. 中国肥厚型心肌病与肌钙蛋白T基因的相关性研究[J]. 南方医科大学学报,2011,31(9):1589-91.
[12]Ripoll-Vera T,Gámez JM,Govea N,et al. Clinical and Prognostic Profiles of Cardiomyopathies Caused by Mutations in the Troponin T Gene[J]. Rev Esp Cardiol (Engl Ed),2016,69(2):149-58.
[13]Gimeno JR,Monserrat L,Pérez-Sánchez I,et al. Hypertrophic cardiomyopathy. A study of the troponin-T gene in 127 Spanish families[J]. Rev Esp Cardiol,2009,62(12):1473-7.
[14]Moolman JC1,Corfield VA,Posen B,et al. Sudden death due to troponin T mutations[J]. J Am Coll Cardiol,1997,29(3):549-55.
[15]Lakdawala NK,Thune JJ,Maron BJ,et al. Electrocardiographic features of sarcomere mutation carriers with and without clinically overt hypertrophic cardiomyopathy[J]. Am J Cardiol,2011,108(11):1606-13.
R542.2
A
1674-4055(2017)11-1386-02
1266041 青岛,青岛市第三人民医院心内科;2100037北京,中国医学科学院阜外医院国家心血管病中心
10.3969/j.issn.1674-4055.2017.11.28
孙竹
