缺氧诱导因子1调控创面微环境的分子机制研究进展*
2017-12-05郝荣涛综述汛审校
陈 兴,郝荣涛,叶 伟 综述,周 汛审校
(重庆市中医院皮肤美容科 400021)
缺氧诱导因子1调控创面微环境的分子机制研究进展*
陈 兴,郝荣涛,叶 伟 综述,周 汛△审校
(重庆市中医院皮肤美容科 400021)
缺氧诱导因子1;创面微环境;分子机制;研究进展
随着社会的发展和人口老龄化的加剧,创面尤其是难愈性创面问题已逐渐成为当今社会不容忽视的一大难题。难愈性创面增加患者经济负担,同时也加大社会医疗资源压力,也严重影响了患者的生存质量。因此,对于创面的相关研究也显得极其重要。
创面愈合是一个较为复杂的生物学过程,包括肉芽组织的形成和创面的再上皮化等基本步骤。在创面愈合过程中,由于创缘及肉芽组织中细胞高耗氧状态,使创面形成一个低氧微环境[1]。有研究指出,慢性难愈性创面周围的持续低氧环境是导致其延迟愈合的重要原因。同时,也有部分研究证实,短时低氧可促进血管的发生及创面的愈合[2]。但短时低氧促进创面愈合及长期低氧延迟创面愈合的具体分子机制目前均未阐明。当前有大量的研究从低氧环境下缺氧诱导因子1(HIF-1)的调控来研究低氧诱导的创面愈合分子机制。本文对当前HIF-1及其对创面调控的分子机制的研究进展进行综述。
1 HIF-1组成及其活性在创面调控中的重要作用
HIF-1是由HIF-1α亚基与HIF-1β亚基共同组成的异二聚体转录因子,其中HIF-1α亚基主要存在于细胞质中,对氧分子及其敏感,而HIF-1β亚基主要存在于细胞核内,只有当HIF-1α亚基进入细胞核后与HIF-1β亚基共同作用才能发挥基因调控作用。由此也可以看出HIF-1的活性主要由HIF-1α亚基决定,这也是HIF-1α是目前HIF-1调控研究关键分子的原因所在。同时也可以看出HIF-1的活性调节主要体现在基因的转录水平和蛋白的表达水平上,例如在特殊情况下增加HIF-1α基因的转录来调控HIF-1的活性,同时也可以反作用调节HIF-1α蛋白的表达[3]。已有大量的研究显示,在创面尤其是慢性创面的愈合过程中,由于其高氧耗极易造成缺氧环境,缺氧环境调控HIF-1对血管内皮生长因子(VEGF)等的表达也起到重要的调控作用[4]。VEGF又叫血管通透因子,是创面愈合过程中的一类重要调控蛋白,对创面及其周围血管的生成起着重要的作用[5],并以此促进创面愈合。除对VEGF的调控外,HIF-1还对创面细胞的增殖及迁移能力起着重要的调控作用。鉴于HIF-1在慢性难愈性创面周围的持续低氧环境及短时低氧环境中的重要作用,HIF-1分子调控机制的探讨对临床创面的预防及治疗均有着极其重要的意义。
2 创面微环境中HIF-1活性调控的分子机制
如前所述,HIF-1α亚基是HIF-1活性调控的关键组成成分,其对氧分子极其敏感,在不同的创面微环境中,其低氧状态调控HIF-1 α亚基从而间接调控HIF-1的活性,并以此影响相关蛋白的表达。慢性创面中,由于组织细胞高耗能,使细胞微环境长期处于低氧状态,此时胞质中HIF-1α亚基感受到低氧刺激并产生活性,随后进入核内与靶基因中的HRE位点(缺氧反应元件)结合。HRE元件通常位于相关靶基因的启动子区域,但也有部分报道指出位于增强子序列中,其核心的关键序列为5′-RCGTG-3′。HIF-1α与该序列结合后,调控下游靶基因的转录,进而调控创面表皮细胞的增殖迁移及创面相关生长因子的表达,并以此促进创面愈合[6];此外,在急性创面中,细胞处于非低(缺)氧的状态,此时HIF-1表达则受到PI3K/Akt/mTOR信号通路及MAPK通路的调控,无论是否缺氧机体均能通过MAKP途径直接磷酸化,从而调控HIF-1的表达。
在急性创面的情况下,创面微环境还未处于低氧状态,此时胞内的脯氨酸羟化酶(PHDs)促进HIF-1α亚基的羟基化,HIF-1α位点羟基化造成泛素化酶的结合,泛素化酶逐渐在细胞质中降解HIF-1α亚基,使其无法入核产生下游效应;而在慢性创面中,细胞微环境处于低氧或缺氧环境中,低氧或缺氧环境促使脯氨酸羟化酶失去活性,此时HIF-1α不被脯氨酸羟化酶羟基化,泛素化酶无法结合,使其产生活性,其后入核调控相关蛋白的表达产生效应,如通过上调脂蛋白受体LRP1影响热休克蛋白HSP90α的表达,从而促进相关细胞的迁移调控创面愈合等[7];也可以通过上调HIF-1α的表达后诱导表皮细胞整合素Integrin β1的表达,以此来促进细胞的迁移及创面愈合[8-9]。总的来看,HIF-1在急性创面及慢性创面中有着不同的分子调控方式。
2.1HIF-1在急性创面或早期创面中的活性调控 细胞通过激活一系列信号通路来应对低氧条件的生存,当前已知的大部分的通路均对HIF-1起到一定的调控,这些调控几乎均依靠对HIF-1的稳定性及转录活性来进行。在急性创面中,细胞微环境还暂时未处于低氧环境,该环境近似为常氧状态,因HIF-1α亚基在急性创面中依旧存在,但由于常氧环境使其极易被羟基化然后被泛素化降解,通常具有不超过5 min的较短寿命,因此HIF-1α亚基在急性创面中也处于一个不断降解与合成的动态过程。需要指出的是在急性创面微环境中,HIF-1α亚基的第402位脯氨酸残基和第564位脯氨酸残基被特异的脯氨酸羟化酶羟基化[10],且此过程需要氧及亚铁离子,及分别以α酮戊二酸及抗坏血酸盐作为辅佐因子参与。这样的催化方式也解释了去铁敏(desferrioxamine)及氯化钴等研究工作中常用的模拟低氧或缺氧铁离子拮抗剂或螯合剂等具有模拟低氧的效应[11]。
戊邻酮二酸盐是三羧酸循环的中间产物,该物质在激活PHDs中扮演着重要的作用,而三羧酸循环中其他产物如延胡索酸等与戊邻酮二酸盐起着拮抗的作用,抑制PHDs的激活[12]。当激活的PHDs与HIF-1α亚基作用后,HIF-1α亚基的第402位脯氨酸残基和第564位脯氨酸残基发生羟基化,VHL蛋白(Hippel-Lindau protein)识别此信息并与其结合。由于VHL蛋白为E3连接酶泛素化蛋白酶复合体重要组成成分,当HIF-1α亚基与VHL蛋白结合后引发VHL蛋白介导的HIF-1α亚基泛素化并逐渐降解[13]。此外,E3连接酶复合体中VHL还连接环指蛋白RBX1,RBX1与同源性的E2蛋白结合,此外VHL蛋白还与其他受体蛋白如延伸因子B(elongin B),延伸因子C(elongin C)及cullin 2等结合形成较大的蛋白复合体[14](图1)。HIF-1α第803位天冬氨酸残基在常氧条件下也容易被命名为FIH-1(factor-inhibiting-1)的天冬氨酸羟化酶羟基化,也阻碍辅助转录蛋白p300/CREB与HIF-1α碳端反式结合区域的结合[15](图1)。
在上述过程中,FIH-1和PHDs蛋白相似,需要在戊邻酮二酸盐、铁离子、抗坏血酸盐及分子氧的共同作用下来促使羟基化,但和PHDs蛋白不同的是,FIH-1蛋白不会因三羧酸循环的中间产物如延胡索酸草酰琥珀酸等而受到抑制。因此,相比HIF-1α第402位脯氨酸残基和第564位脯氨酸残基而言,HIF-1α第803位天冬氨酸残基更容易发生羟基化,影响其活性。HIF-1α亚基在经过上述过程后其活性受到抑制或者逐渐降解,使其无法入核与靶基因HRE元件结合,无法调控下游创面相关基因。因此,HIF-1在急性创面或早期创面的非低氧环境中起到的作用甚微。
急性创面或早期创面微环境还未处于低氧环境,此时HIF-1α第402位脯氨酸残基(402P)及第564位脯氨酸残基(564P)被特异性的PHDs羟基化。VHL蛋白识别HIF-1α脯氨酸残基羟基化位点,并与其结合。VHL蛋白是E3连接酶复合体的重要组成部分,该连接酶复合体还包括elongin B、elongin C、cullin 2及Rbx1。E3连接酶复合体与具有泛素激活活性的E1及泛素连接活性的E2共同作用介导HIF-1α的泛素化,并且逐渐被蛋白酶体降解,以此无法进入细胞核内起调控作用。此外,HIF-1α第803位天冬酰胺残基(803N)被FIH-1识别并羟基化,使其无法与p300/CBP蛋白结合,进一步降低HIF-1α的活性。
2.2HIF-1在慢性创面微环境中的活性调控 在慢性创面以及难愈性创面中,由于细胞一直处于高耗氧,因此其微环境为低氧或缺氧状态,在此时,具有调控HIF-1α亚基羟基化的PHDs及FIH-1在氧分子的作用下均失去活性,HIF-1α亚基的脯氨酸残基均不再发生羟基化,因此HIF-1α也不再发生泛素化降解并在细胞中逐渐累积。当积累到一定程度后,HIF-1α亚基进入细胞核并与HIF-1β形成二聚体结合到相关靶基因的HRE序列(5′-RCGTG-3′五核苷酸序列)上,以此调控细胞迁移、增殖及促进创面愈合的基因表达,促使细胞在低氧状态下生存[16]。此外,在低氧微环境中,p300/CBP蛋白也与HIF-1α相互作用,并抑制HIF-1α亚基第803位天冬氨酸残基的羟基化,以此来增加HIF-1的转录活性[17]。在慢性创面的低氧或缺氧条件下,HIF-1直接或间接调控的基因超过100个,涉及细胞的增殖、分化、迁移及凋亡等[17]。目前研究比较清楚的HIF-1的靶标基因VEGF,对血管的生成具有重要作用,同时也有利于创面的愈合[2](图2)。此外,在众多受HIF-1调控的蛋白中,CXCR4与SDF-1在调控细胞黏附、迁移及在血管的形成中具有极其重要的作用[18](图2)。
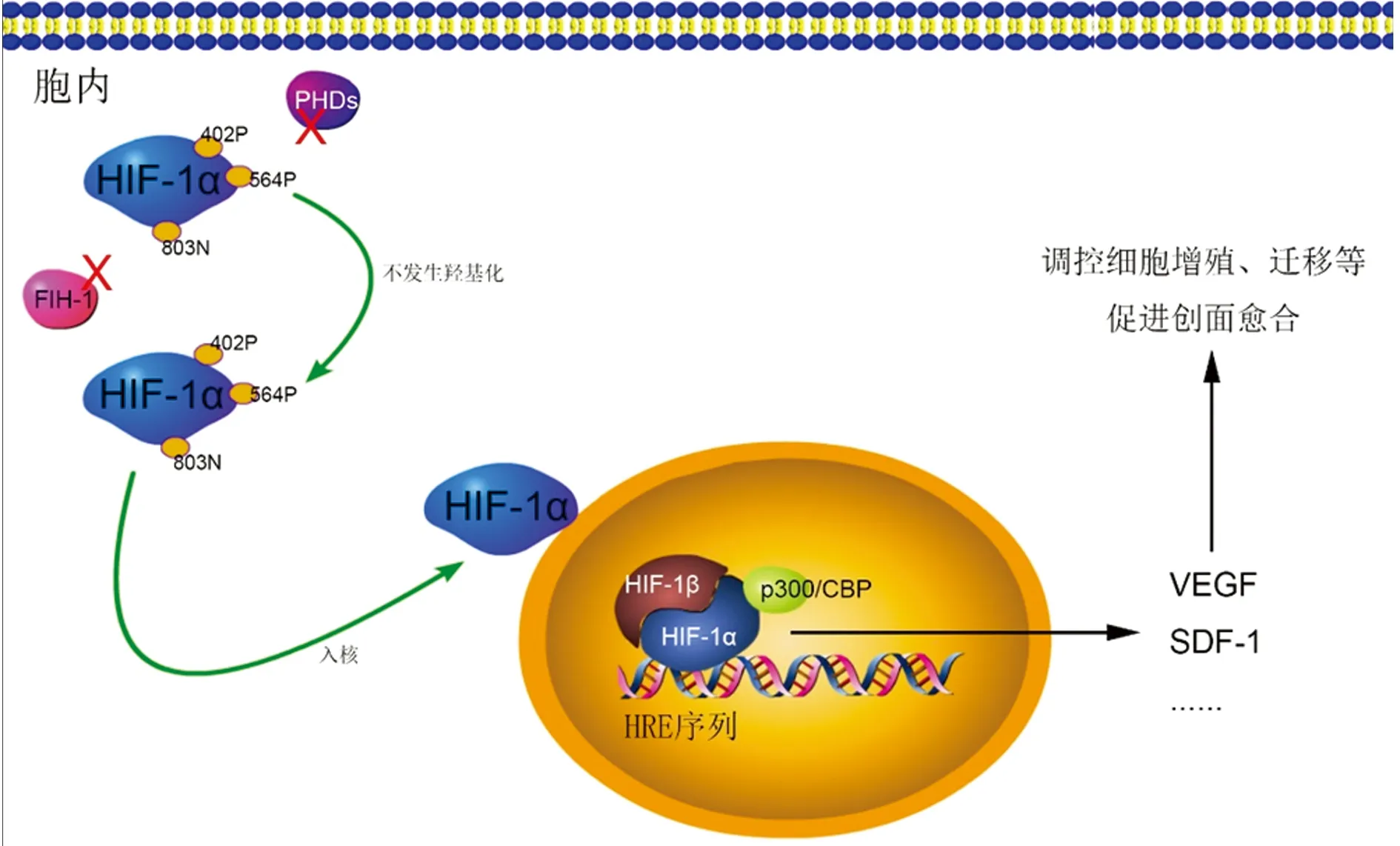
图2 慢性创面微环境中HIF-1α调控模式图
在慢性创面微环境的低氧或缺氧条件下,因PHDs及FIH-1在氧的作用下蛋白失活,使HIF-1α不在羟基化,阻碍其降解。当HIF-1α在细胞质内到达一定量后,HIF-1α进入细胞核内与HIF-1β形成二聚体,并与p300/CBP蛋白相互作用,增强其活性。其后,HIF-1结合到VEGF、SDF-1等靶标基因启动子或增强子区域的缺氧反应元件,促使相关基因转录,以此调控细胞产生应对低氧的反应,调控创面愈合。
2.3HIF-1在创面中的其他调控方式 除上述的早期创面以及慢性创面中的氧微环境对HIF-1活性的调控外,组蛋白脱乙酰基酶抑制剂也介导VHL依靠的HIF-1α蛋白的降解,并以此调控HIF-1活性。组蛋白脱乙酰基酶抑制剂促使HSP90超乙酰化,影响HSP90与HIF-1α的相互作用,形成不成熟的HIF-1α-HSP70复合体,以此影响HIF-1α的活性[19-20]。此外,被命名为CHIP(carboxy terminus of HSP70-interacting protein)的泛素连接酶也对HIF-1α的泛素化及长时间的低氧导致的蛋白的降解过程有着重要的调控作用,同时此过程也在分子伴侣的协助下增加乙二醛的含量[19]。乙二醛同时是糖酵解的侧链产物,与α-酮戊二醛具有较高的亲和性,在慢性创面中具有较高的活性且抑制PDHs的活性,阻碍HIF-1α的羟基化,对HIF-1活性起着重要的调控作用[16]。
尽管已有大量的研究证实HIF-1α的活性调控方式依靠PHD及VHL蛋白促使其降解,但也有证据表明HIF-1α亚基还受其他信号通路的调控,例如RACK1蛋白与HIF-1α蛋白及延伸因子C结合,与起到稳定HIF-1α结构的热激蛋白HSP90竞争性抑制,从而促进HIF-1α泛素化并降解[21]。磷酸化的信号转导通路PI3K及MAPK也对HIF-1α亚基的调控及HIF-1活性对下游相关靶基因的转录及蛋白表达起着调控作用[22-23]。例如,上游正向调节蛋白如络氨酸受体激酶RTKs及小G蛋白Ras的功能获得型突变或者抑癌基因PTEN等肿瘤抑制基因的功能缺失型突变,均能通过PI3K信号通路增强HIF-1α基因的表达[24]。此外,MAPK介导的HIF-1α磷酸化并促使其与p300/CBP蛋白的结合,增强HIF-1的活性[25]。同时HIF-1α磷酸化还能使其不从核内向核外转移,保护其活性。
3 小结与展望
低氧或缺氧是组织在创面尤其是慢性创面中常见的病理生理的反应。大量的研究已经证实,HIF-1α是低氧或缺氧条件下的关键转录调节因子,在促进创面细胞增殖、迁移及创面相关细胞因子的靶基因转录及表达起着重要的作用。HIF-1α活性的调节,也可能是创面尤其是难愈性创面治疗新的切入点。虽然关于HIF-1α活性调节及下游信号对创面愈合的分子调节机制已有较多的认识,但将其准确地应用于临床实践还有很多的问题有待进一步解决。此外,相信随着目前分子生物学研究技术的不断进步,HIF-1α亚基的活性调节也很可能成为除创面外,很多低氧疾病新的靶标,也不断为临床治疗提供新的参考。
[1]Schreml S,Szeimies RM,Prantl L,et al.Oxygen in acute and chronic wound healing[J].Brit J Dermatol,2010,163(2):257-268.
[2]Thangarajah H,Yao DC,Chang EI,et al.The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues[J].Proc Natl Acad Sci USA,2009,106(32):13505-13510.
[3]Wang GL,Jiang BH,Rue EA,et al.Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2tension[J].Proc Natl Acad Sci U S A,1995,92(12):5510-5514.
[4]Bento CF,Pereira P.Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes[J].Diabetologia,2011,54(8):1946-1956.
[5]Dai Y,Xu MF,Wang YG,et al.HIF-1 alpha induced-VEGF overexpression in bone marrow stem cells protects cardiomyocytes against ischemia[J].J Mol Cell Cardiol,2007,42(6):1036-1044.
[6]Barliya T,Mandel M,Livnat T,et al.Degradation of HIF-1 alpha under hypoxia combined with induction of Hsp90 polyubiquitination in cancer cells by hypericin:a unique cancer therapy[J].PloS One,2011,6(9):e22849.
[7]Li W,Li Y,Guan S,et al.Extracellular heat shock protein-90alpha:linking hypoxia to skin cell motility and wound healing[J].EMBO J,2007,26(5):1221-1233.
[8]金秀,王潇飞,王红红,等.硫酸右旋糖苷抑制人胃癌细胞HIF-1α和整合素β1表达及其相关性[J].临床与实验病理学杂志,2016,32(01):53-57.
[9]Lee SH,Lee YJ,Han HJ.Role of hypoxia-induced fibronectin-integrin beta 1 expression in embryonic stem cell proliferation and migration:involvement of PI3K/Akt and FAK[J].J Cell Physiol,2011,226(2):484-493.
[10]Hon WC,Wilson MI,Harlos K,et al.Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL[J].Nature,2002,417(6892):975-978.
[11]Yuan Y,Hilliard G,Ferguson T,et al.Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha[J].J Biol Chem,2003,278(18):15911-15916.
[12]Hewitson KS,Lienard BMR,Mcdonough MA,et al.Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates[J].J Biol Chem,2007,282(5):3293-3301.
[13]Cockman ME,Masson N,Mole DR,et al.Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein[J].J Biol Chem,2000,275(33):25733-25741.
[14]Stebbins CE,Kaelin WG,Pavletich NP.Structure of the VHL-ElonginC-ElonginB complex:implications for VHL tumor suppressor function[J].Science,1999,284(5413):455-461.
[15]Lando D,Peet DJ,Gorman JJ,et al.FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor[J].Genes Dev,2002,16(12):1466-1471.
[16]Weidemann A,Johnson RS.Biology of HIF-1 alpha[J].Cell Death Differ,2008,15(4):621-627.
[17]Sang N,Stiehl DP,Bohensky J,et al.MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300[J].J Biol Chem,2003,278(16):14013-14019.
[18]Ceradini DJ,Kulkarni AR,Callaghan MJ,et al.Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1[J].Nat Med,2004,10(8):858-864.
[19]Bento CF,Fernandes R,Ramalho J,et al.The chaperone-dependent ubiquitin ligase CHIP targets HIF-1 alpha for degradation in the presence of methylglyoxal[J].PloS One,2010,5(11):e15062.
[20]Luo W,Zhong J,Chang R,et al.Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but not HIF-2 alpha[J].J Biol Chem,2010,285(6):3651-3663.
[21]Liu YV,Baek JH,Zhang H,et al.RACK1 competes with HSP90 for binding to HIF-1 alpha and is required for O2-independent and HSP90 inhibitor-induced degradation of HIF-1 alpha[J].Mol Cell,2007,25(2):207-217.
[22]Guo C,Hao LJ,Yang ZH,et al.Deferoxamine-mediated up-regulation of HIF-1 alpha.prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice[J].Exp Neurol,2016,280(1):13-23.
[23]Yeh YH,Wang SW,Yeh YC,et al.Rhapontigenin inhibits TGF-beta-mediated epithelial-mesenchymal transition via the PI3K/AKT/mTOR pathway and is not associated with HIF-1 alpha degradation[J].Oncol Rep,2016,35(5):2887-2895.
[24]Maynard MA,Ohh M.The role of hypoxia-inducible factors in cancer[J].Cell Mol Life Sci,2007,64(16):2170-2180.
[25]Mylonis I,Chachami G,Samiotaki M,et al.Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1 alpha[J].J Biol Chem,2006,281(44):33095-33106.
Q491.5
A
1671-8348(2017)31-4438-04
10.3969/j.issn.1671-8348.2017.31.043
重庆市卫生和计划生育委员会科研项目(20142071)。
陈兴(1986-),住院医师,硕士,主要从事皮肤美容的临床及科研。△
,E-mail:cqzyyzx@163.com。
2017-04-28
2017-07-17)
