HPLC法快速筛查保健食品中非法添加的7种化学药品
2017-12-01连云岚董培智
连云岚,董培智
(山西省食品药品检验所,太原 030001)
HPLC法快速筛查保健食品中非法添加的7种化学药品
连云岚,董培智
(山西省食品药品检验所,太原 030001)
目的 建立用于改善睡眠类保健食品中非法添加卡马西平、氯丙嗪、舒必利、奥卡西平、奥氮平、多赛平和喹硫平7种化学药品的HPLC快速筛查方法。 方法 采用HPLC法,C18柱进行分离(250 mm×4.6 mm;5 μm);检测波长:250 nm;柱温:35 ℃。流动相:甲醇-0.02 mol/L磷酸二氢钾溶液,梯度洗脱。 结果 经对建立的HPLC法进行方法学考察,7种目标分析物的线性范围为20-600 μg/ml,检出限为0.4-2.5 ng/g,定量限为2.5-5.0 ng/g;方法的重复性(RSD)在0.1%-3.6%之间,仪器精密度(RSD)在0.2%-2.8%之间;平均回收率为95-102%;7个成分的溶液在32 h内基本稳定。 结论 该HPLC法操作简便,能同时快速筛查保健食品中卡马西平等7种非法添加化学药品。
改善睡眠类保健食品; HPLC; 非法添加化学药品
由于生活节奏加快,工作压力加大,社会中失眠患者日益增多,借助药物来改善睡眠的人群也在不断增加。不法分子为了达到快速治疗失眠的效果,在保健食品中非法添加镇静催眠类化学药物。现有的国家局颁布的3个补充检验方法[1-3]收录的改善睡眠类非法添加的化学药物共有22种,但根据近年来我所检测近500批次的结果显示未发现有添加上述药物。除上述22种药物之外,还有一些具有睡眠作用的精神类药物也可能添加到中成药和保健食品中,此类药物往往会造成相当严重的不良反应,而且具有耐药性和依赖性,大多被列为国家二类精神药品。如卡马西平具有嗜睡作用,但其为抗惊厥、三叉神经痛药,并有抗抑郁及抗心律失常的作用[4-5];氯丙嗪为抗精神病药,具有神经安定作用,长期使用会产生耐药性[6]。因此有必要对改善睡眠类保健食品中非法添加的其他精神类药品进行检测方法的研究。本实验建立了HPLC(检测器采用DAD)时检测睡眠类保健食品中卡马西平、氯丙嗪、舒必利、奥卡西平、奥氮平、多赛平和喹硫平7种非法添加的精神类药品的方法。
1 仪器和试药
1.1 仪器
戴安U3000高效液相色谱仪,MS-204s万分之一电子天平,DV215CD十万分之一电子天平,超声波提取仪。
1.2 试剂及对照品
磷酸二氢钾,分析纯;甲醇,分析纯(提取)、色谱纯(流动相);盐酸,分析纯。对照品均来源于中国食品药品检定研究院:卡马西平(批号:100142-20110S,规格:100 mg,含量99.8%;富马酸喹硫平(批号:100815-201202,规格:100 mg,含量99.9%);盐酸氯丙嗪(批号:100460-201302,规格:100 mg,含量99.7%);奥氮平(批号:100948-200801,规格:100 mg,含量99.9%);盐酸多塞平(批号:100069-201103,规格:100 mg,含量99.5%);奥卡西平(批号:100657-201102,规格:100 mg,含量99.8%);舒必利(批号:100203-200503,规格:100 mg,含量100%)。
31批次样品来源于2015年全国保健食品监督抽检任务辅助睡眠类保健食品中不含褪黑素的样品。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent C18柱(4.6 mm×250 mm,5 μm);柱温:室温35 ℃。流动相:甲醇-0.02 mol/L磷酸二氢钾溶液,梯度洗脱见表1。检测波长:250 nm。流速:1.0 ml/min。
表1梯度洗脱表
Table1Gradientelutiontable

时间(min)A(甲醇)B(磷酸二氢钾)0-555%-95%95%-5%55-6495% 5% 65-755% 95%
2.2 供试品制备
精密称取次服用量至50 ml容量瓶中,加甲醇40 ml,超声提取15 min,放至室温,用甲醇定容至刻度。0.45 μm膜过滤,取续滤液,作为样品待测液备用。
2.3 标准曲线的制备
精密称定上述7种对照品每种约12 mg分别置于25 ml容量瓶中,卡马西平、富马酸喹硫平、盐酸多塞平、米氮平4种对照品用约20 ml的甲醇溶解,奥卡西平、舒必利、奥氮平3种对照品用约20 ml的乙腈溶解。用溶媒定容后,摇匀,作为储备液备用。
精密量取对照品储备液各1.0 ml至同一个10 ml量瓶中,用甲醇稀释至刻度,摇匀,制成每1 ml含对照品约50 μg的对照品溶液。然后取5.0 ml定容至10.0 ml,摇匀,配制成不同浓度的对照品溶液作为标准曲线。以进样量(μg)为横坐标,积分面积为纵坐标求得回归方程,结果线性范围在20-600 μg/ml之间(见表2)。
2.4 稳定性试验
对照品储备液配制后冰箱冷藏保存,测试溶液在配制后不同时间测定其峰面积,计算其RD,对对照品测试溶液进行稳定性考察。结果各指标在32 h内基本稳定(见表3)。
表27种指标线性范围
Table2Thecalibrationcurvesof7chemicaldrugsandtheirranges
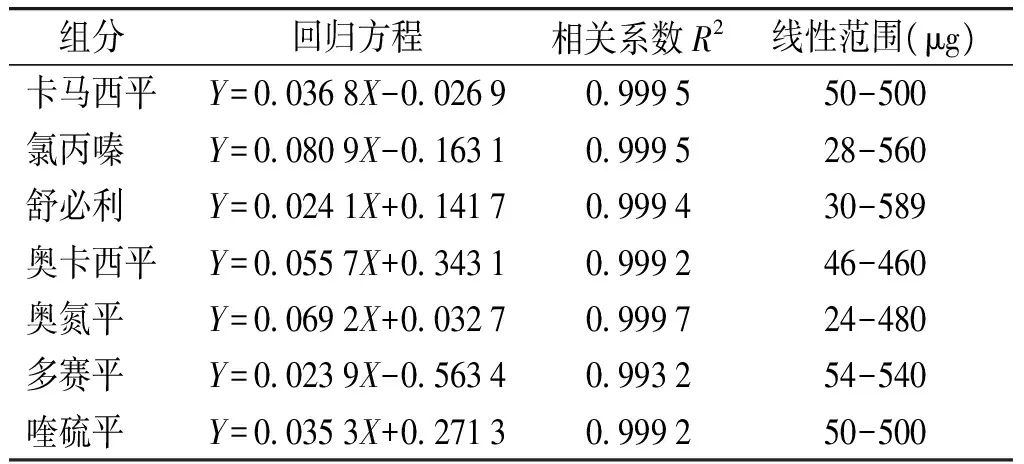
组分回归方程相关系数R2线性范围(μg)卡马西平Y=00368X-002690999550-500氯丙嗪 Y=00809X-016310999528-560舒必利 Y=00241X+014170999430-589奥卡西平Y=00557X+034310999246-460奥氮平 Y=00692X+003270999724-480多赛平 Y=00239X-056340993254-540喹硫平 Y=00353X+027130999250-500
表3稳定性实验结果(n=10)
Table3Stabilitytestresult(n=10)
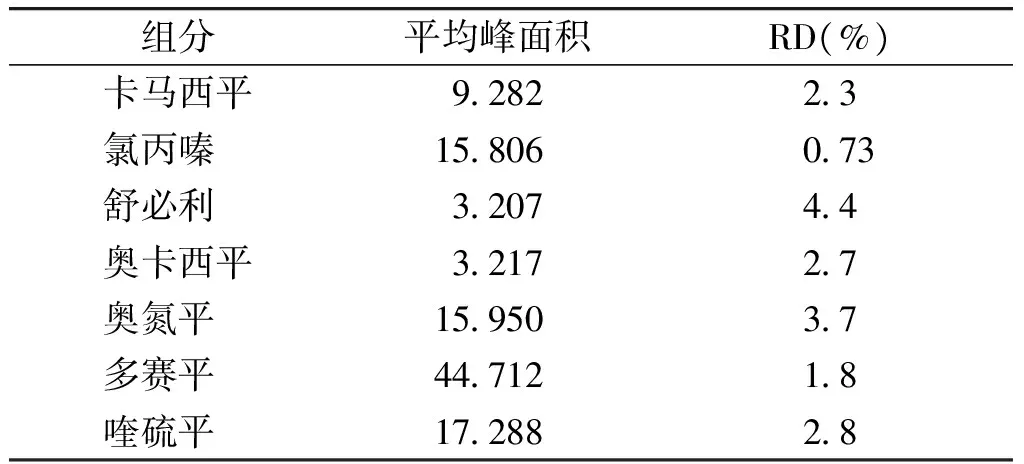
组分平均峰面积RD(%)卡马西平928223氯丙嗪 15806073舒必利 320744奥卡西平321727奥氮平 1595037多赛平 4471218喹硫平 1728828
2.5 检测波长的选择
考虑到不同化学药品最大吸收波长不同(分别为卡马西平237 nm;氯丙嗪256 nm;舒必利242 nm;奥卡西平256 nm;奥氮平257 nm;多赛平256 nm;喹硫平256 nm),选择250 nm为其大多数物质吸收值较高的波长。
2.6 仪器精密度考察
选择标准曲线中间浓度的对照品溶液连续进样5次,计算峰面积的RSD,结果仪器精密度RSD小于5%(见表4)。
2.7 重复性实验
用加标样替代样品进行重复性试验。分别取1 ml对照品溶液于5个已称样品的10 ml容量瓶,甲醇定容至刻度。进样10 μl,测定样品中各组分的量。测定结果表明:重复性试验符合要求(见表5)。
2.8 准确度
采用加样回收法测定准确度。精密量取某口服液1.0 ml分别至9个10 ml容量瓶中,分别精密加入浓度为0.469 5 mg/ml的对照品溶液1 ml,0.8 ml,0.4 ml各3份,加甲醇至大约8 ml,超声15 min提取,定容至10 ml。取续滤液,进样10 μl,计算回收率,结果回收率在95%-102%之间(见表6)。
表4仪器精密度试验
Table4Theprecisiontestofinstrument
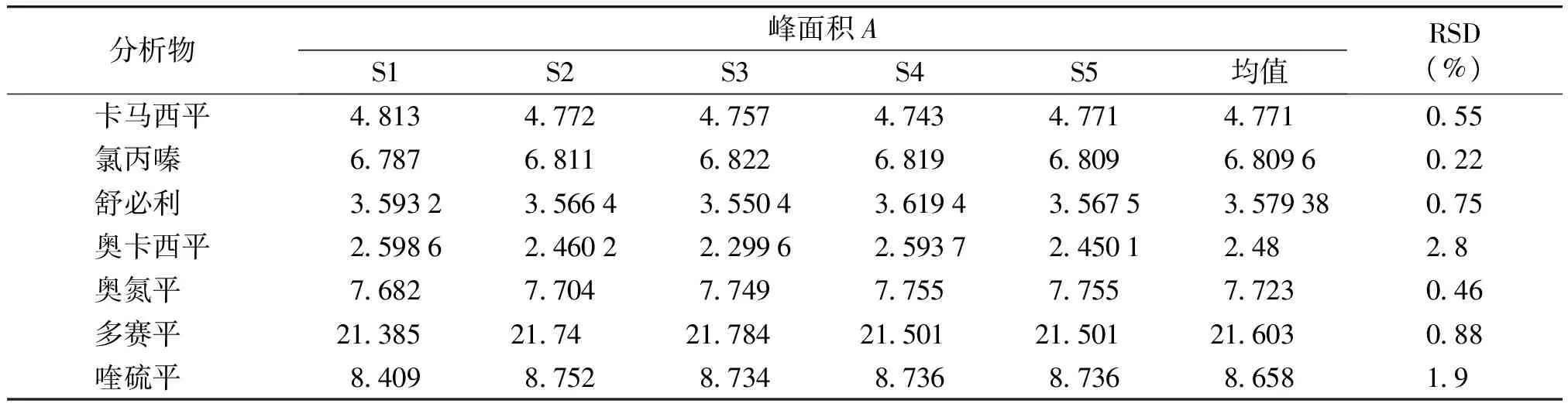
分析物峰面积AS1S2S3S4S5均值RSD(%)卡马西平481347724757474347714771055氯丙嗪 6787681168226819680968096022舒必利 3593235664355043619435675357938075奥卡西平259862460222996259372450124828奥氮平 768277047749775577557723046多赛平 21385217421784215012150121603088喹硫平 84098752873487368736865819
表57种对照品重复性试验
Table5Repeatabilitytestof7kindsofchemicaldrugs
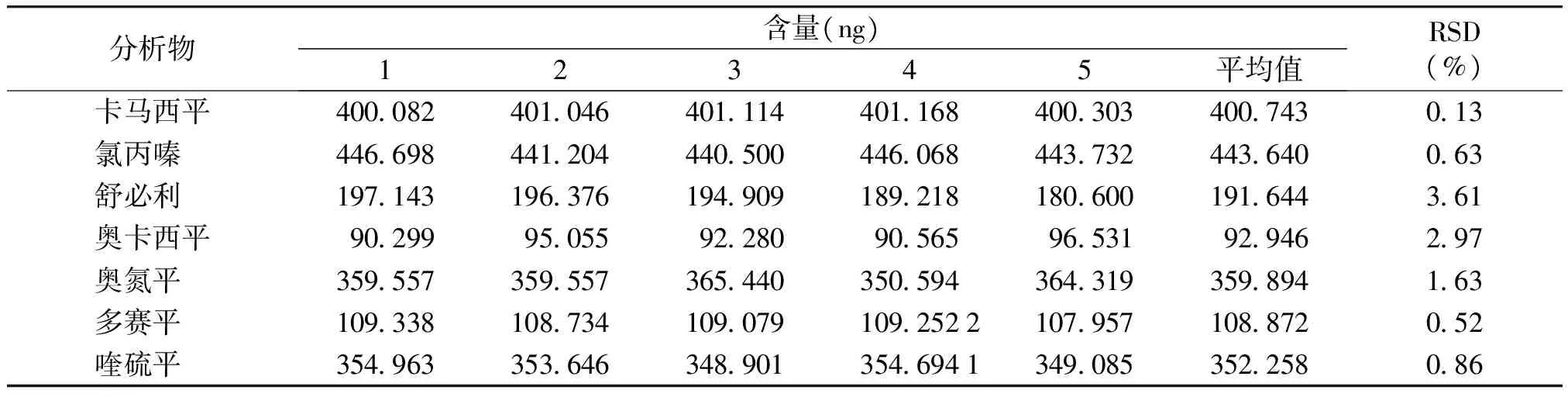
分析物含量(ng)12345平均值RSD(%)卡马西平400082401046401114401168400303400743013氯丙嗪 446698441204440500446068443732443640063舒必利 197143196376194909189218180600191644361奥卡西平902999505592280905659653192946297奥氮平 359557359557365440350594364319359894163多赛平 1093381087341090791092522107957108872052喹硫平 3549633536463489013546941349085352258086
表67种对照品平均回收率
Table6Theaveragerecoveriesof7kindsofchemicaldrugs
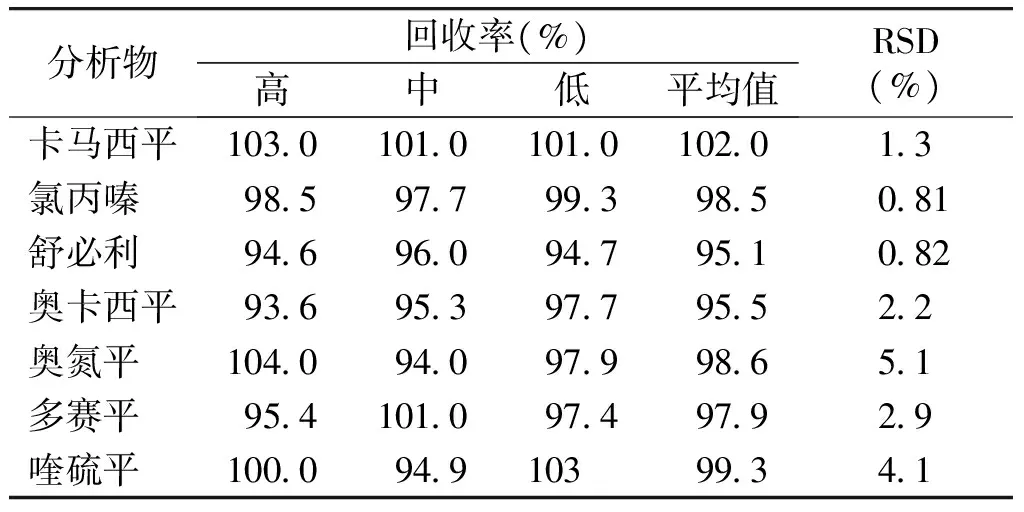
分析物回收率(%)高中低平均值RSD(%)卡马西平103010101010102013氯丙嗪 985977993985081舒必利 946960947951082奥卡西平93695397795522奥氮平 104094097998651多赛平 954101097497929喹硫平 100094910399341
2.9 检出限、定量限
将加标样品溶液逐级稀释,过滤后进样,以峰高信噪比10 ∶1的对照品浓度为定量浓度,带入称样量及稀释体积后折算定量限(见表7);同法以峰高信噪比3 ∶1的对照品浓度为检出浓度,折算检出限。结果检出限在0.4-2.5 ng/g之间,定量限在2.5-5.0 ng/g之间(见表7)。
2.10 样品测定
根据验证的方法,对本次任务中31批次样品各10 μl进行测定,结果均未检出有非法添加(见图1)。
3 讨论
本文为促进睡眠类保健食品中非法添加物初筛方法研究。供试品溶液中不得出现与对照品溶液保留时间一致的色谱峰,如果出现保留时间一致的色谱峰,则其光谱图应不一致,如色谱保留时间、光谱图均与对照品一致,则用液质联用仪测定,进行干扰排除。对供试品溶液中出现与对照品溶液保留时间、光谱、质谱均一致的色谱峰,则判定为有非法添加,用以上方法进行定量。以峰面积为纵坐标,以不同进样浓度为横坐标,做标曲曲线,将样品峰面积带入,计算样品待测液浓度。
表7检出限与定量限
Table7Thedetectionlimitsandthequantitativelimitsofof7kindsofchemicaldrugs

成分检出限(ng/g)定量限(ng/g)卡马西平04925氯丙嗪 05628舒必利 05829奥卡西平04627奥氮平 04824多赛平 05427喹硫平 25050
本项目7个非法添加物的选择是参照文献[9]筛选出9种具有睡眠作用的精神类药物,其中米氮平和盐酸西普肽兰因不稳定,故未列入本次方法学考察。
实验中以甲醇-磷酸二氢钾、乙腈-磷酸二氢钾为流动相进行梯度洗脱考察,发现以甲醇-磷酸二酸钾梯度洗脱时,色谱峰分离度好,实验效果最好。故最终选择甲醇-磷酸二酸钾梯度洗脱。
[1] 国家食品药品监督管理总局.安神类中成药和保健食品中非法添加褪黑素、佐匹克隆、氯苯那敏、扎来普隆的补充检验方法.药品检验补充检验方法和检验项目批准件(批准件编号2012004)[S].[2012-08-02].
[2] 国家食品药品监督管理总局.安神类中成药中非法添加化学品检测方法.药品检验补充检验方法和检验项目批准件(批准件编号2009024)[S].[2009-12-04].
[3] 国家食品药品监督管理总局.改善睡眠类中成药及保健食品中非法添加罗痛定、青藤碱、文拉法辛补充检验方法.药品检验补充检验方法和检验项目批准件(批准件编号2013002)[S].[2013-03-26].
[4] 曹冬冬,李丙英,王守双.卡马西平片的含量测定方法[J].江苏药学与临床研究,2003,11(3):16-18.
[5] 付萍萍,景浩然,赵志伟,等.高效液相色谱法测定复方卡钙菖蒲胶囊中卡马西平的含量[J].时珍国医国药,2007,1(5):1151-1152.
[6] 徐景晖,丁海芳,项志通,等.HPLC法测定氯丙嗪、异丙嗪的含量[J].中国药师,2008,11(2):243-244.
[7] 张圆,闵春艳,郝刚,等.保健食品中非法添加化学药物的检测方法[J].中国职业药师,2014,11(7):35-39.
[8] 国家药典委员会.中国药典:2010年版四部[S].北京:中国医药科技出版社,2015:374.
[9] 罗疆南,谭力,曹玲,等.LC-MS/MS法同时测定中药制剂中非法添加的15种抗癫痫药及镇静催眠类药物[J].中国生化药物杂志,2010,31(3):22-26.
Rapiddeterminationof7kindsofillegally-addedchemicaldrugsinhealthfoodbyhighperformanceliquidchromatography
LIAN Yunlan,DONG Peizhi
(ShanxiInstituteforFoodandDrugControl,Taiyuan030001,China)
ObjectiveTo establish a rapid screening method for the simultaneous determination of 7 kinds of illegally-added chemical drugs, including Carbamazepine, Chlorpromazine, Sulpiride, Oxcarbazepine, Olanzapine, Doxepin and Quetiapine, in improving sleep quality health food by high performance liquid chromatography(HPLC).MethodsA C18column (250 mm×4.6 mm,5 μm) was used for chromatographic separation, which was eluted with methanol-0.02 mol/L potassium dihydrogen phosphate as mobile phase at 35 ℃.The diode array detector was used for detection at 250 nm.ResultsThe 7 chemical drugs showed a linear relationship in the range of 20-600 μg/ml. The limit of determination and the limit of quantification of the method were in the ranges of 0.4-2.5 ng/g and 2.5-5.0 ng/g,respectively. The RSD of repeatability was in the range of 0.1%-3.6% and the precision of instrument was 0.2%-2.8%.The average recovery rate was 95%-102%. The above seven components solution was stable in 32 h.ConclusionThis method is simple and convenient, and suitable for simultaneous rapid determination of 7 kinds of illegally-added chemical drugs in health food.
improving sleep quality health food; high performance liquid chromatography; illegally-added chemical drugs
连云岚,女,1972-10生,硕士,副主任药师,E-mail:lianyunlan2006@163.com
2017-07-21
R927.2
A
1007-6611(2017)11-1165-04
10.13753/j.issn.1007-6611.2017.11.016
