基于质谱技术的贝伐珠单抗及其糖基化修饰的表征、定量与药代动力学分析
2017-11-06丛宇婷胡良海叶明亮顾景凯邹汉法
丛宇婷 胡良海 叶明亮 顾景凯 邹汉法
1(吉林大学生命科学学院,长春 130012)2(中国科学院大连化学物理研究所,中国科学院分离分析化学重点实验室,大连116023)
基于质谱技术的贝伐珠单抗及其糖基化修饰的表征、定量与药代动力学分析
丛宇婷1,2胡良海*1叶明亮2顾景凯1邹汉法2
1(吉林大学生命科学学院,长春 130012)2(中国科学院大连化学物理研究所,中国科学院分离分析化学重点实验室,大连116023)
利用基质辅助激光解析电离飞行时间质谱技术、SDS-聚丙烯酰胺凝胶电泳技术以及Shot-gun蛋白质组学策略对贝伐珠单克隆抗体药物及其糖基化修饰进行了全面表征,共发现17种糖型并确定了特征肽段序列。利用液相色谱-串联质谱联用技术,采用平行反应检测模式,通过测定单抗水解后产生的特征肽段,实现了对大鼠血浆样品中单抗药物含量的绝对定量分析,获得不同给药计量下的贝伐珠单抗的药物浓度-时间曲线,同时实现了对单抗糖基化修饰各糖型的相对定量分析。本实验首先建立标准工作曲线,对大鼠血浆样品中的贝伐珠单抗进行定量分析,在线性范围内,相关系数R2=0.998,定量限为66 fmol,线性关系良好。对大鼠血浆样品的测定结果表明,高、低计量给药的贝伐珠单抗药物浓度-时间曲线趋势基本一致,浓度均为直接下降,与理论趋势相符。但是大多数糖型呈现浓度先上升的现象,之后的代谢情况因糖型差异而有所不同。
贝伐珠单抗; 糖基化修饰; 液相色谱-串联质谱联用; 平行反应监测; 药代动力学
2017-03-20收稿;2017-09-07接受
本文系国家自然科学基金项目 (No. 81373374)资助
1 引 言
治疗性单克隆抗体药物(Therapeutic monoclonal antibody, mAb; 以下简称单抗药物)是一类以γ型免疫球蛋白G为结构基础的大分子蛋白类药物,为多种疾病的治疗提供了全新的途径[1,2]。贝伐珠单抗是一种重组的人源化单克隆抗体药物,可选择性地与人血管内皮生长因子(Vascular endothelial growth factor, VEGF)结合并阻断其生物活性,用于治疗直肠癌[3]。贝伐珠单抗药物的序列已知,分子量约为149 kDa,其中轻链分子量约为23 kDa,重链分子量约为49 kDa。作为复杂的大分子糖蛋白药物,单抗药物两条重链的Fc片段上297位的天冬氨酸(Kabat编号Asn297)上具有一个高度保守的N-糖基化修饰位点[4]。其N-糖基化修饰通常是复杂的双天线糖链结构,可能发生岩藻糖化和唾液酸化。大量研究表明,单抗药物的N-糖基化修饰对其稳定性、清除率、免疫原性、抗体依赖细胞毒性(Antibody-dependent cellular cytotoxicity, ADCC)及补体依赖细胞毒性(Complement-dependent cytotoxicity, CDC)等都有一定的影响[5]。例如,缺少核心岩藻糖的糖型能够增强对FcγRIIIа受体的亲和力,从而提高ADCC[6]; 而高水平的唾液酸化的糖型则会减少对FcγRIIIа受体的亲和力,从而降低ADCC[7]。因此,建立稳定可靠的分析方法,进行单抗药物及其糖基化修饰的药代动力学及药效动力学研究具有重要意义。
传统的蛋白类药物的药代动力学研究多采用酶联免疫吸附方法(Enzyme-linked immunosorbent assays, ELISA)[8,9]。尽管该技术为蛋白定量分析提供了一种简单、灵敏、高通量的方法,但仍存在很多局限性,如研制周期长、成本高、无法区分内源性干扰以及存在交叉反应等,并且难以用于对翻译后修饰的研究[10,11]。近年来,随着质谱技术的不断完善与发展,液相色谱-串联质谱联用技术(Liquid chromatography-tandem mass spectrometry,LC-MS/MS)逐步成为蛋白质定量的高灵敏度、高特异性、高通量以及重现性好的分析方法,其中多反应监测(Multiple reaction monitoring,MRM)技术通过两次离子选择,极大地降低了背景噪声,从而提高了分析方法的灵敏度和重现性,已成为蛋白质定量的重要手段[12]。此外,MRM方法在糖基化修饰定量研究中也展现出极大优势。Toyama等[13]建立了一种通过“能量依赖型”氧鎓离子监测模式对单抗药物的N-糖型进行定量表征的方法,系统研究了质谱能量与N-糖链氧鎓离子碎裂情况之间的关系,实现了对N-糖链的定量表征,糖肽的检出限为30 amol,线性范围达4个数量级,并可区分糖链的同分异构体。该研究为单抗药物的N-糖型的定量研究提供了新思路。Hong等[14]利用MRM策略同时对IgG蛋白及其N-糖型进行绝对定量分析,IgG蛋白的检出限为60 amol,检测动态范围达3个数量级,对IgG蛋白进行绝对定量分析的同时,可在不经过富集的情况下直接对血清中26种高丰度N-糖肽进行测定。近年出现的四极杆-静电场轨道阱(Q-Orbitrap)高分辨质谱仪提供了另一个类似MRM的选择—平行反应监测(Parallel reaction monitoring,PRM)模式。PRM与MRM的不同之处在于,第3个四极杆被高分辨率、高质量精度的Orbitrap质量分析器代替,它可在单次分析中检测所有的碎片离子,因此称为平行反应监测[12,15,16]。尽管PRM策略已成功用于蛋白定量研究中[17,18],但目前尚无PRM策略用于单抗药物分析的相关报道。
本研究基于PRM技术,建立了一种可同时对单抗药物及其糖基化修饰的定性与定量质谱分析方法,并应用于药代动力学研究中。本研究为蛋白类药物的药代动力学研究提供了新方法,为糖蛋白药物的糖基化修饰研究提供了新思路,并且为药物研发和临床安全用药的研究提供了新的途径和理论依据。
2 实验部分
2.1仪器与试剂
AB Sciex 5800 MALDI-TOF/TOF质谱仪(美国AB SCIEX公司); Protein IEF Cell垂直电泳仪(美国Bio-Rid公司); Thermo Q-Exactive质谱仪(美国Thermo公司),配有纳升级电喷雾离子源,Xcalibur 2.1数据处理软件以及Thermo Ultimate 3000超高效液相色谱系统; 恒温振荡器、离心浓缩仪(美国Thermo公司); UMAX PowerLook2100xl扫描仪(中国力广科技股份有限公司)。
贝伐珠单抗(100 mg/4 mL)购自本地药店; 稳定同位素标记的特征肽段FTFSLDTSK*(*所示为同位素13C,15N标记的赖氨酸)购自北京中科亚光生物科技有限公司; 肽糖苷酶F(PNGase F,美国New England Biolabs公司); 尿素(Urea,加拿大Bio Basic公司); 乙腈(ACN,色谱纯,德国Merck公司); 胰蛋白酶、二硫苏糖醇(DTT)、碘乙酰胺(IAA)、4-羟乙基哌嗪乙磺酸(HEPES)、过硫酸铵(AP)、3,5-二甲氧基-4-羟基肉桂酸(SA)、甲酸(FA)和三氟乙酸(TFA)均购自美国Sigma公司; 四甲基乙二胺(TEMED,美国Fluka公司); 还原SDS-PAGE供试品缓冲液(5×)、40%丙烯酰胺溶液(美国Thermo公司); 考马斯亮蓝G250(美国Bio-Rad公司); 固相萃取柱(10 mg,美国Waters公司); 实验用水为超纯水Milli-Q系统(美国Milipore公司)制备的超纯水。
2.2色谱与质谱条件
实验室自制毛细管C18trap柱(5 cm×200 μm); 自制电喷雾喷针的C18色谱柱(15 cm×75 μm); 流动相A:H2O-FA(100∶0.1,V/V); 流动相B:ACN-H2O-FA(80∶20∶0.1,V/V); 梯度洗脱: 0~10 min,96% A; 10~11 min,96%~92% A; 11~100 min,92%~55% A; 100~105 min,55~10% A; 105~113 min,10% A; 113~115 min,10~96% A; 115~130 min,96% A。流速:300 nL/min。
正离子反应模式; 平行反应监测模式; 1次全扫加10次目标扫描为1个循环; 全扫扫描范围为m/z400~2500,分辨率为70000,自动增益设为1×106,最大注入时间为50 ms; 目标扫描分离窗口为±1, 分辨率为35000,自动增益设为1×106,最大注入时间为200 ms。质谱参数见表1。
2.3数据处理
酶解考察:用Proteome Discoverer 1.3软件(美国Thermo公司)将Q-Exactive质谱仪采集生成的*.raw文件转成*.mgf文件,然后通过Mascot Daemon 2.3.0搜索引擎(英国Matrix Science公司)进行数据库检索,数据库为自建贝伐珠单抗数据库,参数设置如下:质量容忍度母离子为10 ppm,碎片离子为0.05 Da; 肽段需符合胰蛋白酶半酶切限制,最多允许两个漏切位点; 半胱氨酸设为固定修饰(Carboxyamidomethylation,57.0215); 甲硫氨酸设为可变修饰(Oxidized methionine,15.9949 Da); 导出肽段有效阈值p<0.01,打分值Score>10,最终获得*.csv搜库结果文件。
糖基化修饰表征:将去糖基化样品的*.raw文件采用上述方式通过贝伐珠单抗数据库进行数据检索得到*.csv文件,可变修饰设置中增加天冬酰胺脱酰胺化(Deamidated asparagine,0.9840 Da),其它
表1 贝伐珠单抗特征肽段、内标序列及其糖基化位点所在肽段EEQYN*STYR对应的各糖肽在PRM模式下的质谱条件
Table 1 Sequences of selected specific peptide and internal standard peptide for bevacizumab and constitutions of its glycoforms of peptide EEQYN*STYR and their parameters for parallel reaction monitoring (PRM) analysis
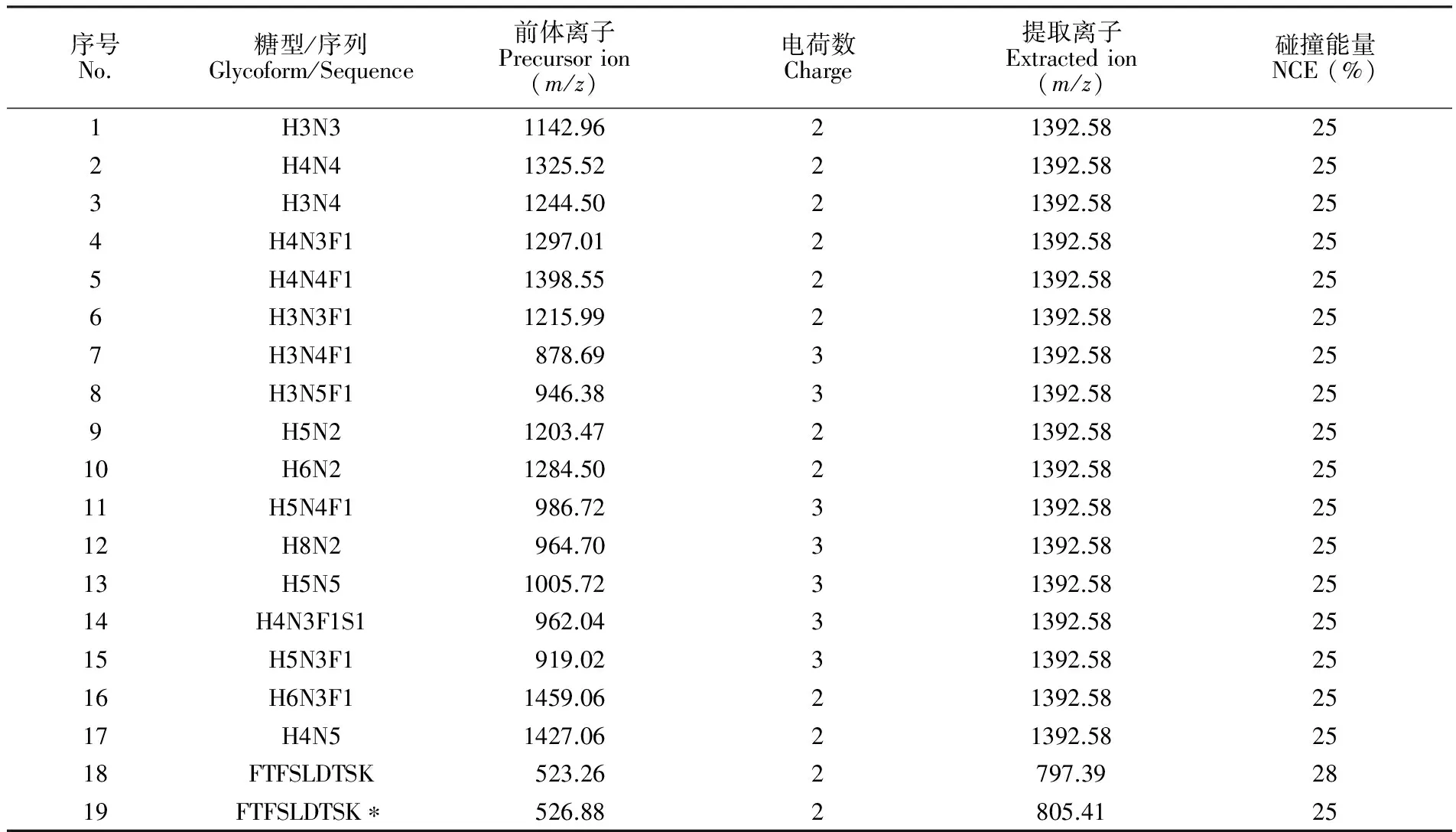
序号No.糖型/序列Glycoform/Sequence前体离子Precursorion(m/z)电荷数Charge提取离子Extractedion(m/z)碰撞能量NCE(%)1H3N31142.9621392.58252H4N41325.5221392.58253H3N41244.5021392.58254H4N3F11297.0121392.58255H4N4F11398.5521392.58256H3N3F11215.9921392.58257H3N4F1878.6931392.58258H3N5F1946.3831392.58259H5N21203.4721392.582510H6N21284.5021392.582511H5N4F1986.7231392.582512H8N2964.7031392.582513H5N51005.7231392.582514H4N3F1S1962.0431392.582515H5N3F1919.0231392.582516H6N3F11459.0621392.582517H4N51427.0621392.582518FTFSLDTSK523.262797.392819FTFSLDTSK∗526.882805.4125
相同。然后通过ArMone 2.0软件将*.csv文件转为*.ppl文件。同时将贝伐珠单抗酶解样品的*.raw文件通过MS Convert软件转为*.mzXML文件。最后,将生成的*.ppl文件与*.mzXML文件一同导入ArMone软件,进行自动搜库匹配,获得糖基化修饰结果。
2.4给药方案及血浆样品采集
取雄性大鼠两只,重量为(200±20) g,自由饮食、饮水,分别按照50和10 mg/kg的高、低剂量进行尾部静脉推注给药。
采取大鼠眼眶静脉丛采血方式,对两组剂量大鼠分别按给药前(0 h)和给药后0.5、1、2、4、8、12、24、32 h采集血液1 mL于肝素化1.5 mL离心管中, 4℃ 15000 r/min离心5 min,然后于80℃冷冻保存,待测。
2.5贝伐珠单抗标准系列溶液及内标肽段溶液的配制
取适量贝伐珠单抗储备液(25 μg/μL),用水分别稀释得到贝伐珠单抗标准系列溶液,浓度为1.2、1.0、0.5、0.1、0.04、0.01和0.005 μg/μL,于20℃保存备用。
取适量内标肽段储备液(5.0 μg/μL),用水稀释配制成终浓度为0.01 μg/μL的内标肽段溶液。
2.6标准曲线样品的制备
分别取4 μL贝伐珠单抗标准系列溶液置于2 mL离心管中,加入4 μL大鼠空白血浆、188 μL变性缓冲液(50 mmol/L HEPES,8 mol/L Urea,pH 8.0)、4 μL DTT(1 mol/L),涡旋混匀,60℃恒温振荡1 h。加入1.5 mg固体IAA,涡旋混匀,避光,25℃恒温振荡40 min。加入1.4 mL缓冲液(50 mmol/L HEPES,pH 8.0),涡旋混合,再加入6 μL胰蛋白酶溶液(2 μg/μL),涡旋混合,37℃恒温振荡15 h。加入10 μL FA溶液,终止反应。加入20 μL 内标肽段溶液(0.01 μg/μL),涡旋混匀。将上述得到的酶解液通过固相萃取去除盐等杂质,用1.2 mL ACN-H2O-TFA(80∶20∶0.1,V/V)溶液洗脱,收集洗脱液,离心,冻干。加入200 μL 0.1% FA溶液复溶,得到标准曲线样品,取2 μL进样进行质谱分析。
2.7大鼠血浆样品的酶解
分别取4 μL各时间点大鼠血浆样品置于2 mL 离心管中,加入192 μL变性缓冲液(50 mmol/L HEPES, 8 mol/L Urea, pH 8.0), 4 μL 1 mol/L DTT, 涡旋混匀, 60℃恒温振荡1 h。 其余步骤同上。
3 结果与讨论
3.1单抗序列和糖基化分析
基于Top-down的蛋白质组学策略,对贝伐珠单抗及其糖基化修饰进行了完整表征。首先,通过MALDI-TOF MS技术,在完整蛋白水平对贝伐珠单抗的分子量及糖基化修饰情况进行了检测。如图1A所示,测得的完整贝伐珠单抗蛋白的分子量均约为150 kDa,与理论值相符。此外,谱图中在76 kDa的峰为单抗的二价分子离子峰。去糖基化的贝伐珠单抗蛋白的测定结果如图1B所示,测得的去糖基化贝伐珠单抗蛋白的分子量均约为147 kDa,与理论值相符。图1B中在74 kDa处同样有一个峰,同样为单抗的二价离子峰。去糖基化后的蛋白及片段的分子量均比未经过去糖基化的低2~4 kDa,这与单抗药物常见糖基化的几种糖型分子量一致[5,13,19]。
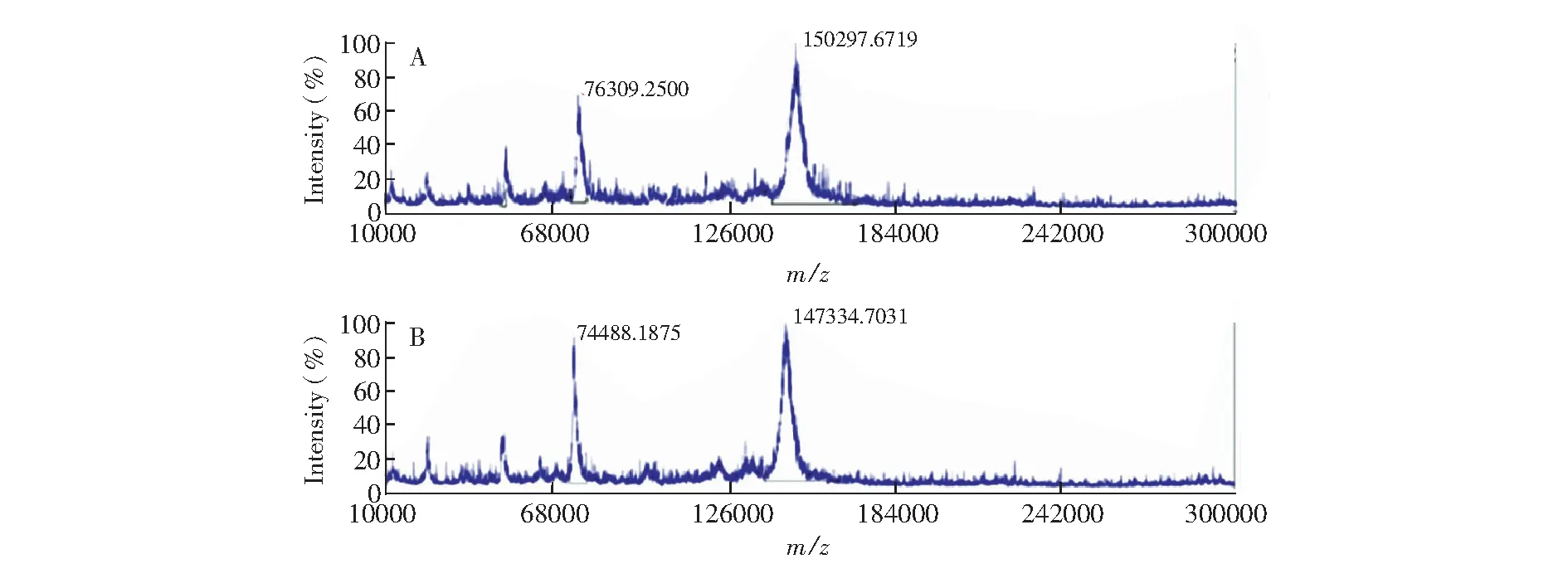
图1 贝伐珠单抗的MALDI谱图(A)完整贝伐珠单抗; (B)去糖基化贝伐珠单抗Fig.1 MALDI-TOF-MS spectra of (A) intact bevacizumab and (B) deglycosylated bevacizumab

图2 贝伐珠单抗SDS-PAGE电泳图。M: 蛋白质marker; A: 贝伐珠单抗; B: 去糖基化贝伐珠单抗; C: 贝伐珠单抗轻重链; D: 去糖基化贝伐珠单抗轻重链Fig.2 Sodiumdodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) map of anti-bevacizu monoclonal (bevacizumab). M: protein marker; A: bevacizumab; B: deglycosylated bevacizumab; C: light chains and heavy chains of bevacizumab; D, light chains and heavy chains of deglycosylated bevacizumab
同时,利用SDS-PAGE技术,对贝伐珠单抗完整蛋白及其糖基化修饰和贝伐珠单抗轻重链及其糖基化修饰进行了表征。在进行完整蛋白的表征时,为了保持蛋白完整性,采取变性非还原SDS-PAGE对其进行考察,即选择非还原上样缓冲液; 而在对轻重链进行表征时,选择还原上样缓冲液,通过缓冲液中的DTT将二硫键打开。SDS-PAGE电泳结果如图2所示,A、B分别为基于变性非还原电泳的完整贝伐珠单抗及其去糖基化条带,仔细观察可以发现,A条带略宽于B条带,这主要是由于糖基化修饰导致的微观不均一性引起的。此外,B条带略靠前,这是由于去糖基化后分子量减小的缘故,但是由于减少的部分只占完整蛋白分子量中比例很小,因此条带距离差异并不明显。条带C、D分别为基于变性还原电泳的贝伐珠单抗轻重链及其去糖基化的条带。C和D中均在50及25 kDa附近有条带,对应其重链和轻链,与理论结果一致。C和D中的轻链条带几乎无差别,贝伐珠单抗的轻链并没有N-糖基化修饰,因此无论是否经过去糖基化处理,轻链的分子量应该无明显差别,显示结果与理论相符。C和D中的重链结果差别明显,C中重链的条带明显宽于D中的,并且D中重链的条带明显比C更靠前,这均与糖基化修饰相关。糖基化修饰导致的微观不均一性使C中重链的条带更宽,而去糖基化后分子量减小使D中重链的条带更靠前。而相对于完整单抗蛋白而言,糖基化修饰占重链的比例更大,因此C、D中重链之间的距离比A、B中条带的距离更大。
采用高分辨质谱技术对贝伐珠单抗药物的胰蛋白酶酶解肽段进行考察,结果见图3A; 通过数据库检索获得酶解肽段信息,结果如图3B和3C所示,轻链序列覆盖率98%,重链序列覆盖率86%。没有覆盖的肽段主要是由于长度或者氨基酸残基缺失等原因导致难以检测。
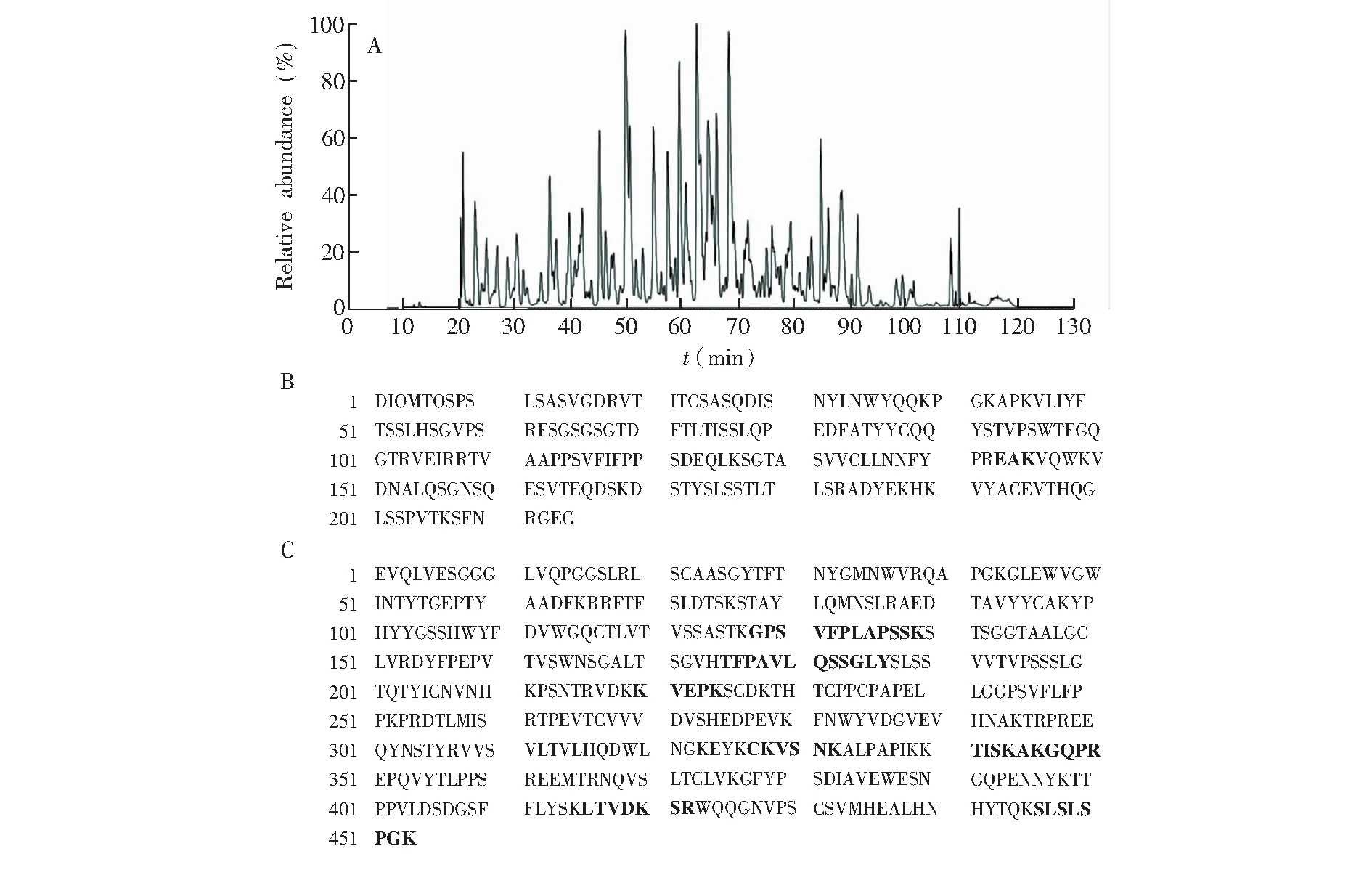
图3 贝伐珠单抗胰蛋白酶酶解:(A)色谱图; (B)轻链覆盖率(98%); (C)重链覆盖率(86%)Fig.3 (A) Chromatogram of trypsin digestion of bevacizumab; Coverage of (B) light chain (98%) and (C) heavy chain (86%). Bold sequences are matched sequences
同时,结合胰蛋白酶酶解、PNGase F去糖基化和ArMone搜库策略,对贝伐珠单抗的N-糖基化修饰进行了全面表征。PNGase F酶可将N-链接糖链从天冬酰胺脱落,同时天冬酰胺被转化为天冬氨酸,使肽段分子量增加0.98 Da,从而实现对糖基化修饰位点的检测[20]。ArMone是一个高通量的完整N-糖肽分析软件平台,用于糖基化修饰位点以及糖链结构的分析。该软件通过结合去糖基化的肽段检测得到的糖基化位点所在肽段信息以及完整糖肽的检测得到糖链结构信息,最终给出完整糖肽的序列、糖链结构以及糖基化位点信息[21]。通过该策略确定单抗的糖基化位点位于重链的第303位天冬氨酸残基(*所示),所在肽段序列为EEQYN*STYR。共表征到图4所示的17种糖型。
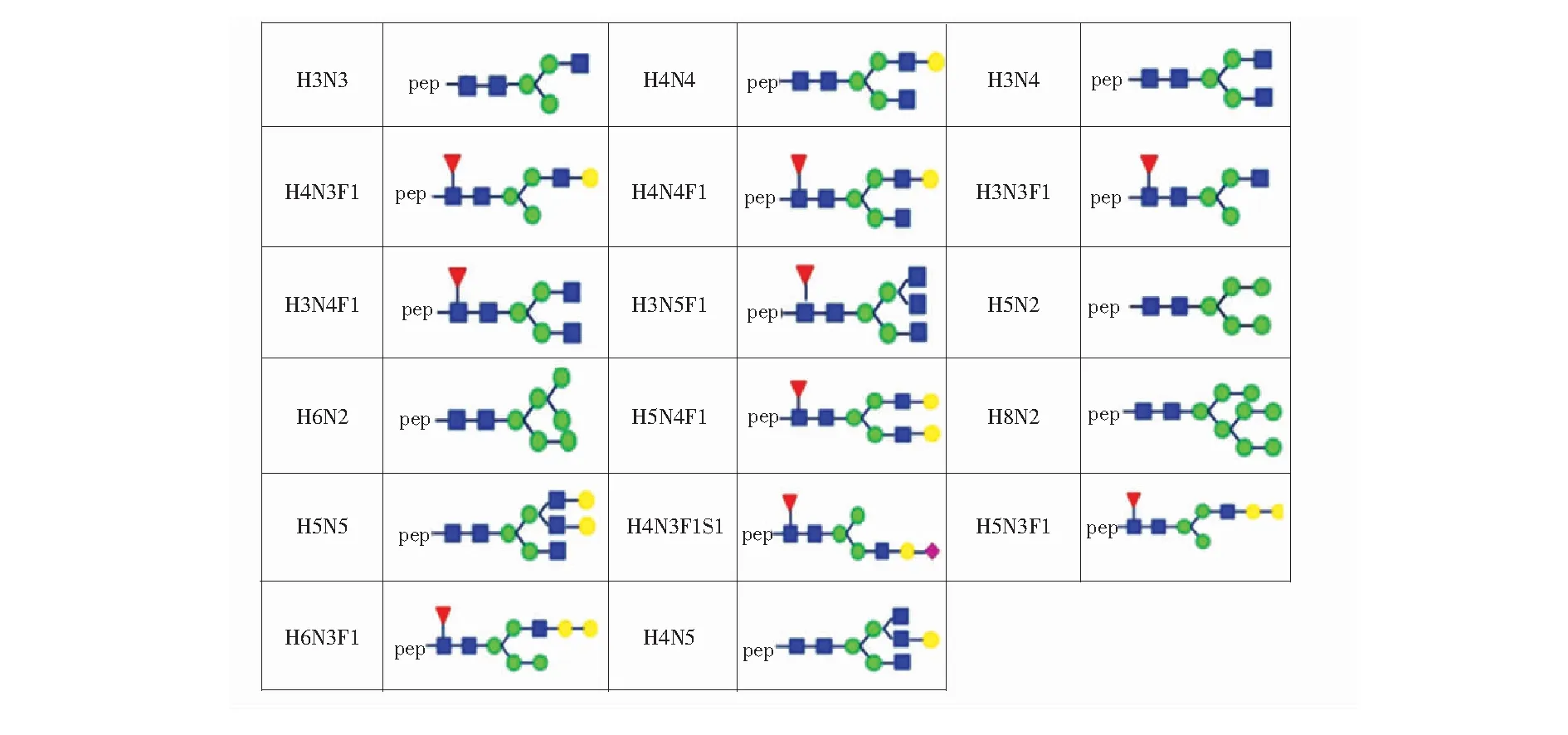
图4 贝伐珠单抗17种糖型的结构示意图Fig.4 Structure diagram of 17 kinds of glycoforms of bevacizumab N-乙酰葡糖胺(H); 甘露糖(N) ; 半乳糖(N) ; 岩藻糖(F); 唾液酸(S)。 N-Acetyl glucosamine; Mannose; Galactose; Fucose; Sialic acid.
3.2单抗药物及其糖基化修饰的药代动力学分析
特征肽段的选择是建立可靠的、重现性好的定量方法的基础,根据特征肽段的筛选原则,本研究采用理论筛选与实际检测相结合策略进行特征肽段的筛选,最终确定贝伐珠单抗的特征肽段序列为FTFSLDTSK(重链的68~86位,分子量为1044.5129 Da)。根据对大鼠实际样品的预实验结果确定标准曲线的浓度范围,通过对一系列不同浓度的贝伐珠单抗进行酶解,同时加入等量的空白血浆同步酶解提供基质环境,并引入合成的稳定同位素标记的肽段做内标,建立贝伐珠单抗的大鼠血浆标准曲线。以待测物(即特征肽段)在血浆中的浓度为横坐标,待测物色谱图峰面积与内标肽段色谱图峰面积的比值为纵坐标,用加权最小二乘法进行回归运算,得到的线性回归方程即为大鼠血浆标准曲线,R2=0.998,定量限为33 fmol/μL,最低在柱样品量为1.3 fmol,线性关系良好。
本研究分别采用10和50 mg/kg两个剂量对大鼠进行静脉推注给药,通过标准曲线测得各采血时间点的血浆药物浓度,得到高、低剂量贝伐珠单抗的药物浓度-时间曲线(图5)。两个剂量下的贝伐珠单抗药物浓度-时间曲线趋势基本一致,浓度均是直接下降,与静脉推注给药的理论趋势一致。此外,在低剂量实验中,当浓度降为0.2 mg/mL时有一个明显的延缓趋势,而降到0.1 mg/mL时再次出现平缓降低的趋势,并且之后一直保持比较平缓的下降趋势。高剂量实验中,浓度降到0.5 mg/mL时出现了延缓趋势,浓度降到0.2 mg/mL时同样出现了一个延缓趋势,此结果与低剂量实验一致。
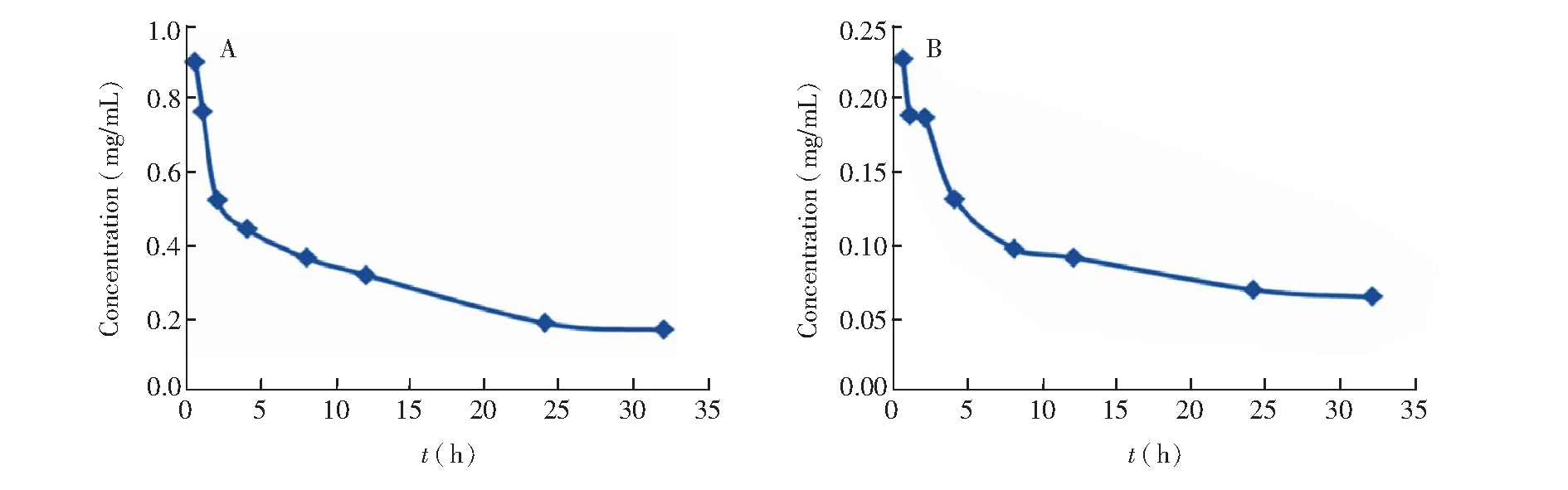
图5 贝伐珠单抗鼠血浆药物浓度-时间曲线(A)高剂量(50 mg/kg)实验; (B)低剂量(10 mg/kg)实验Fig.5 Blood concentration-time curves of bevacizumab after administration of a single intravenous (A) 50 mg/kg and (B) 10 mg/kg dose with rats
采用此方法在对贝伐珠单抗药物进行绝对定量分析的同时,可实现对其糖基化修饰的相对定量分析。以峰面积代表其相对含量,得到高、低剂量大鼠给药后血浆中17种糖基化修饰的变化情况。结果表明(图6),在高剂量给药大鼠血浆中,所有糖型均能检测到,其中H3N3、H4N4、H3N4、H4N3F1、H4N4F1、H3N3F1、H3N4F1、H3N5F1、H5N2、H6N2和H5N4F1等糖型在整体上均是先上升后下降,在下降过程中会出现短暂的平缓趋势,然后再次下降时会有延缓趋势。H8N2、H5N5、H4N3F1S1和H5N3F1等糖型在整体上的变化情况比较一致,均是先上升,不同的是,在第一次下降之后,均出现了至少一次的再次上升趋势。H6N3F1和H4N5两种糖型,均是先下降然后出现了两次显著上升的情况。而低剂量给药大鼠中,四种糖基化修饰的血浆浓度随时间变化趋势与高剂量吻合,对比结果如图6所示,其余的糖型则因为含量低而难以分析整体变化趋势。在糖肽的变化过程中,2 h内普遍存在上升趋势,这与单抗药物本身的变化过程不一致,目前尚无文献报道该现象,推测可能是糖基化影响了单抗药物在体内的转运。
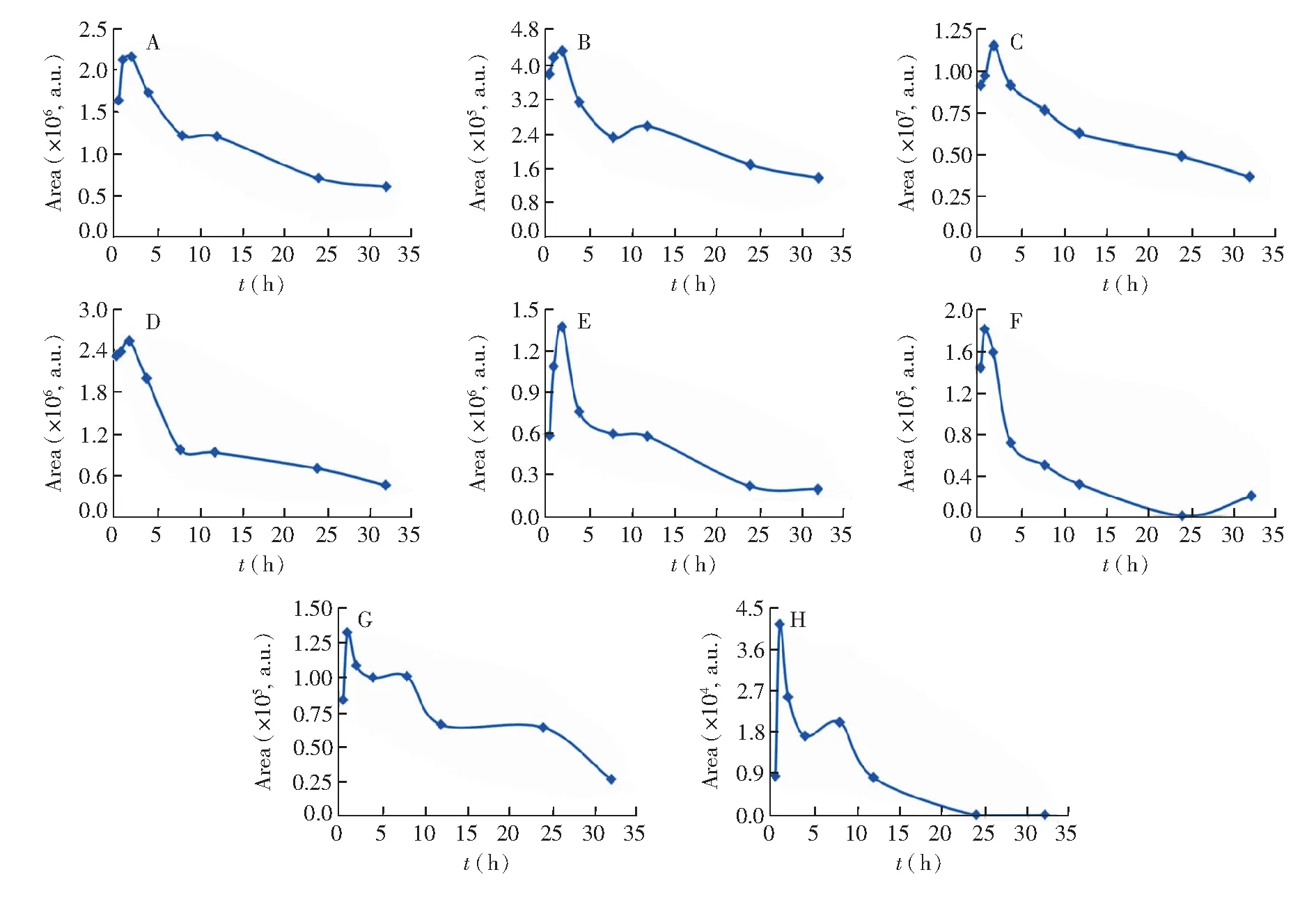
图6 贝伐珠单抗4种糖基化修饰的血浆浓度-时间曲线高低剂量对比结果: A: 50 mg/kg H4N4F1; B: 10 mg/kg H4N4F1; C: 50 mg/kg H3N4F1; D: 10 mg/kg H3N4F1; E: 50 mg/kg H5N2; F: 10 mg/kg H5N2; G: 50 mg/kg H5N4F1; H: 10 mg/kg H5N4F1Fig.6 Comparison results of blood concentration-time curves of 4 glycoforms of bevacizumab after administration of a single intravenous 50 mg/kg and 10 mg/kg dose with rats. A: 50 mg/kg H4N4F1; B: 10 mg/kg H4N4F1; C: 50 mg/kg H3N4F1; D: 10 mg/kg H3N4F1; E: 50 mg/kg H5N2; F: 10 mg/kg H5N2; G: 50 mg/kg H5N4F1; H: 10 mg/kg H5N4F1
4 结 论
本研究基于LC-MS/MS 技术,建立了一种对贝伐珠单抗药物及其糖基化修饰的PRM体内定量分析方法,此方法简单、可靠、灵敏度高、通量高,能同时实现对单抗药物本身及其糖基化修饰的定量分析,为单抗药物的药代动力学研究提供了一种新方法。
1 Chen Z, Wang J, Bao L, Guo L, Zhang W, Xue Y, Zhou H, Xiao Y, Wang J, Wu F, Deng Y, Qin C, Jin Q.Nat.Commun.,2015, 6: 6714
2 Hogarth P M, Pietersz G A.Nat.Rev.DrugDiscov.,2012, 11(4): 311-331
3 Al-Debasi T, Al-Bekairy A, Al-Katheri A, Al Harbi S, Mansour M.SaudiJ.Ophthalmol.,2017, 31(2): 99-105
4 Reusch D, Haberger M, Maier B, Maier M, Kloseck R, Zimmermann B, Hook M, Szabo Z, Tep S, Wegstein J, Alt N, Bulau P, Wuhrer M.MAbs,2015, 7(1): 167-179
5 Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianferani S.Anal.Chem.,2013, 85(2): 715-736
6 Chung S, Quarmby V, Gao X,Ying Y, Lin L, Reed C, Fong C, Lau W, Qiu Z J, Shen A, Vanderlaan M, Song A.MAbs,2012, 4(3): 326-340
7 Scallon B J,Tam S H, McCarthy S G, Cai A N, Raju T S.Mol.Immunol.,2007, 44(7): 1524-1534
8 Engvall E, Perlmann P.Immunochemistry,1971, 8(9): 871-874
9 Gan S D, Patel K R.J.Invest.Dermatol.,2013, 133(9): e12
10 Mesmin C, Fenaille F, Ezan E, Becher F.Bioanalysis,2011, 3(5): 477-480
11 Ezan E, Bitsch F.Bioanalysis,2009, 1(8): 1375-1388
12 Ronsein G E, Pamir N, von Haller P D, Kim D S, Oda M N, Jarvik G P, Vaisar T, Heinecke J W.J.Proteomics,2015, 113: 388-399
13 Toyama A, Nakagawa H, Matsuda K, Sato TA, Nakamura Y, Ueda K.Anal.Chem.,2012, 84(22): 9655-9662
14 Hong Q, Lebrilla C B, Miyamoto S, Ruhaak L R.Anal.Chem.,2013, 85(18): 8585-8593
15 Lesur A, Domon B.Proteomics,2015, 15(5-6): 880-890
16 Tsuchiya H, Tanaka K, Saeki Y.Biochem.Biophys.Res.Commun.,2013, 436(2): 223-229
17 Urisman A, Levin R S, Gordan J D, Webber J T, Hernandez H, Ishihama Y, Shokat K M, Burlingame A L.Mol.CellProteomics,2017, 16(2): 265-277
18 Sundberg M, Strage E M, Bergquist J, Holst B S, Ramström M.PLosOne,2016, 11(12): e0167138
19 Siemiatkoski J, Lyubarskaya Y, Houde D, Tep S, Mhatre R.Carbohydr.Res.,2006, 341(3): 410-419
20 Bobaly B, D'Atri V, Goyon A, Colas O, Beck A, Fekete S, Guillarme D.J.Chromatogr.B,2017, 1060: 325-335
21 Cheng K, Chen R, Seebun D, Ye M, Figeys D, Zou H.J.Proteomics,2014, 110: 145-154
This work was supported by the National Natural Science Foundation of China (No. 81373374).
Characterization,QuantificationandPharmacokineticAnalysisof
BevacizumabandItsGlycosylationbyMassSpectrometry
CONG Yu-Ting1,2, HU Liang-Hai*1, YE Ming-Liang2, GU Jing-Kai1, ZOU Han-Fa2
1(CollegeofLifeScience,JilinUniversity,Changchun130012,China)
2(CASKeyLaboratoryofSeparationSciencesforAnalyticalChemistry,NationalChromatographicR&ACenter,
DalianInstituteofChemicalPhysics,ChineseAcademyofSciences,Dalian116023,China)
The bevacizumab and its glycoforms were analyzed by matrix-assisted laser desorption ionization-time of flight-time of flight-mass spectrometry (MALDI-TOF-MS), sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and short-gun strategy, with the sequence of unique peptide and seventeen glycoforms being characterized. The bevacizumab and its glycopeptides concentrations in mice plasma with different intravenous injection doses of bevacizumab were detected and the concentration-time curves were obtained by parallel reaction monitoring (PRM) method based on liquid chromatography-tandem mass spectrometry (LC-MS/MS) technique. First, standard curves were created for quantification of mAb in mice plasma, which showed good linearity, with the correlation coefficient (R2) value of 0.998 and the lower limit of quantification of 66 fmol. Detection results of high and low doses of the drug in the mice plasma samples showed that the drug concentration-time curve trend was consistent, e.g. the concentration was decreasing. However, the results of quantitation of seventeen glycoforms demonstrated that the metabolism of different glycoforms was different. The concentrations of most glycoforms increased first, whereas the metabolism afterwards differed by different glycoforms.
Bevacizumab; Glycosylation; Liquid chromatography-tandem mass spectrometry; Parallel reaction monitoring; Pharmacokinetics
20 March 2017; accepted 7 September 2017)
10.11895/j.issn.0253-3820.170170
