J波综合征发生机制的研究进展
2017-10-20宋焕秋刘元生
宋焕秋 刘元生
·心血管急诊·
J波综合征发生机制的研究进展
宋焕秋 刘元生
目的 J波综合征是引起自发性室颤导致心脏性猝死的一个重要原因。虽然J波综合征有多种表型,但其发生机制与心电图表现具有相似性。J波是动作电位1相复极的结果,主要由Ito介导的瞬时外向钾电流产生。由于心内膜与心外膜Ito通道密度不同,构成了J波产生的生理基础。除了Ito外,其它离子通道如INa、Ica、Ik也参与J波的形成,这提示任何一种离子通道异常都可产生J波。其中,最常见的离子通道病变是基因突变。已有大量实验表明J波综合征与多种离子通道基因突变相关,如SCN5A、CACNA1C、CACNB2、SCN1B、KCNE3。这些突变的离子通道影响心外膜复极,产生2相折返而引发室颤。因此,充分了解J波综合征的发生机制,有助于临床诊断和治疗,降低死亡率。
J波综合征; ERS; BrS; 离子通道; 基因突变
J波综合征是指心电图上有J波出现的临床症候群,包含多种表型,具有发生室颤等心脏猝死性心律失常的风险。J波综合征包括早复极综合征(ERS)、Brudaga综合征(BrS)、先天性室颤(IVF)伴下壁导联(Ⅱ、Ⅲ、aVF)J波。已有研究证明,J波综合征不同分型之间的心电图表现有差异,但其产生J波的离子与细胞机制以及危险因素是相同的[1-2]。本文针对J波综合征相关离子、分子以及基因学病理机制进行简要综述。
一、J波综合征的离子通道机制
1.J波综合征与瞬时外向钾通道(Ito):心电图上记录的电活动是对心肌细胞膜离子通道动态变化所产生的动作电位的一种描述,这些离子通道激活使细胞膜去极化,随后通道恢复稳态,细胞膜复极,这两种变化相对应于心肌细胞的收缩与舒张过程。心肌细胞膜动作电位由五部分组成,即0~4相。0相为除极相,由快钠通道介导的内向钠离子电流(INa)产生;1~4相为复极相,涉及多种离子通道,主要由钾通道所产生的外向钾离子流(IK)与钙通道内向钙离子流(ICa)。1相连接除极与复极,在心电图上的表现就是J波,因此J波为心肌细胞除极与复极的转折点,心电图上相对应的位置是紧随QRS波之后的一正向波[3]。
1相的快速复极主要是Ito所产生的外向钾离子流的作用。此通道为快通道,激活与失活时间短,对膜电位变化敏感[4]。心脏的除极顺序是从心内膜到心外膜,而复极顺序则相反,由心外膜到心内膜。实验证明心外膜Ito通道的密度大于心内膜,因此在除极过程中从心外膜到心内膜产生了电压梯度,这在心电图上就表现为J波,因此J波是心外膜1相除极的结果[5]。凡影响Ito通道性能以及心室除极顺序的因素,都可导致J波的产生。除此之外,钠通道与L型钙通道异常引起内向INa与Ica电流减弱以及其他外向Ik电流增强也会影响J波[6-8]。在正常心脏,从心外膜到心内膜的正常顺序除极将J波隐藏于QRS波之中,因此心电图上无J波出现。有时J波不能被完全遮盖,在心电图上表现为QRS波尾端的顿挫或拖尾,类似预激波形态[9](图1)。J波的产生与Ito通道的状态密切相关,因此心率可明显影响J波的产生,心率减缓时J波明显,幅度增大,心率加快时J波幅度减小甚至消失[10]。
作者单位:100044 北京大学人民医院急诊科
2.2相折返机制引发室颤:如果1相Ito电流使心外膜动作电位切迹足够深时,以至于达到L型钙通道开放阈值,使心外膜动作电位圆顶可能完全消失。但在心外膜动作电位圆顶消失之前首先表现出圆顶出现延迟,从而引起动作电位时程延长,呈现下斜型ST段抬高,这其实是一巨大的J波,并伴有T波倒置(图2)。随后,心外膜动作电位圆顶完全消失,动作电位时程缩短。但心外膜动作电位圆顶的消失并非一致,一些区域的圆顶消失,另一些区域圆顶还处于延迟阶段,使跨室壁复极离散度增强[11-12]。由于不同区域动作电位时程不同,圆顶消失区域复极速度快,类似于早后除极(EAD)发生机理,因此在电压差作用下低电位区域可产生一个新的动作电位,形成短-偶联异位搏动,从而引发室颤。心外膜动作电位圆顶形成于2相时期,因此称为2相折返机制[13]。
二、J波综合征相关基因突变
1.Brudaga综合征心电图特点及突变基因:Brudaga综合征的心电图特点是J波增高伴有右侧胸导联(V1~V3)完全或不完全性右束支传导阻滞(通常称为拱形ST段抬高)以及T波倒置,拱形抬高形态的ST段具有很高心脏猝死率[14-15]。Brudaga综合征是一种常染色体遗传病,包括12个基因突变缺陷,这些突变导致多种离子通道功能异常,包括Ito、IL-ca、INa、Ik(表 1)。
2.早复极综合征心电图特点及突变基因:早复极综合征(ERS)心电图特点是出现明显的J波伴有R波尾端顿挫或拖尾,ST段抬高。根据不同导联的表现,将ERS分为3种亚型:Ⅰ型:早复极形态心电图出现在侧壁导联,常见于健康运动员;Ⅱ型:早复极形态心电图出现在下侧壁导联,可见于正常人,也可见于先天性室颤患者;Ⅲ型:早复极形态心电图出现在下侧壁以及右侧胸导联,具有发展为恶性心律失常的高风险[15]。ERS也存在基因突变,相关突变基因有6个(表2)。
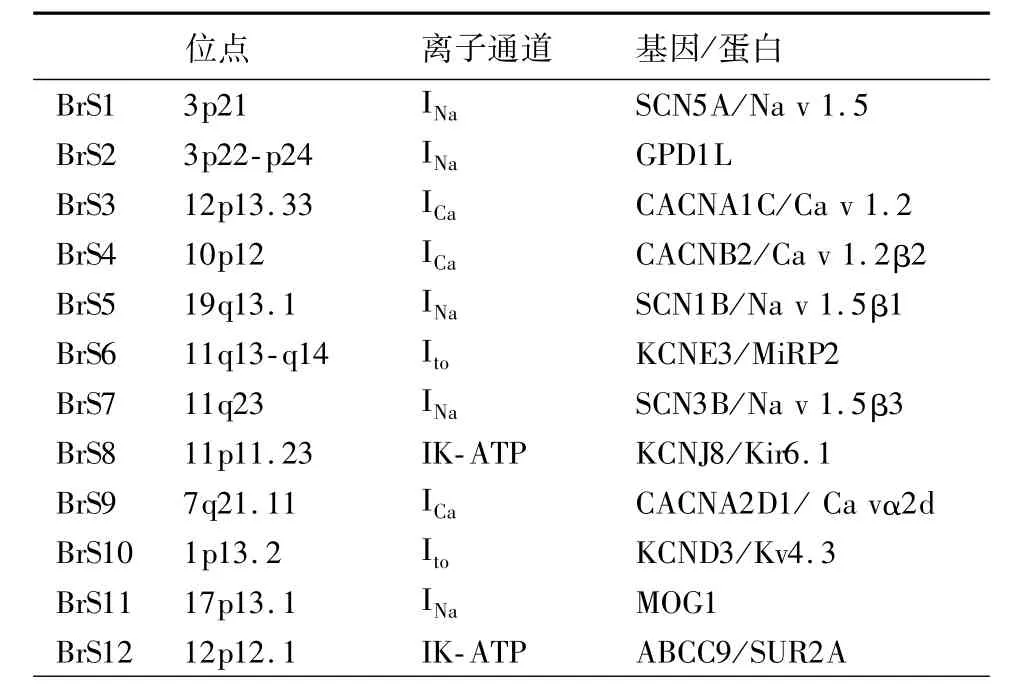
表1 BrS相关突变基因
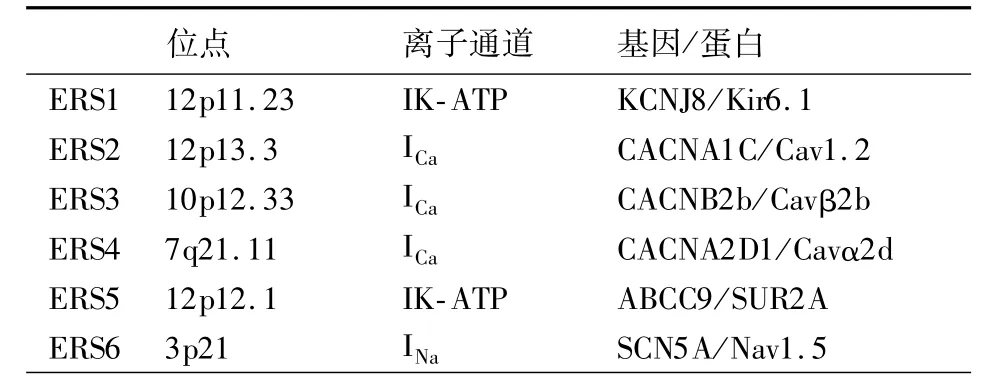
表2 ERS相关突变基因
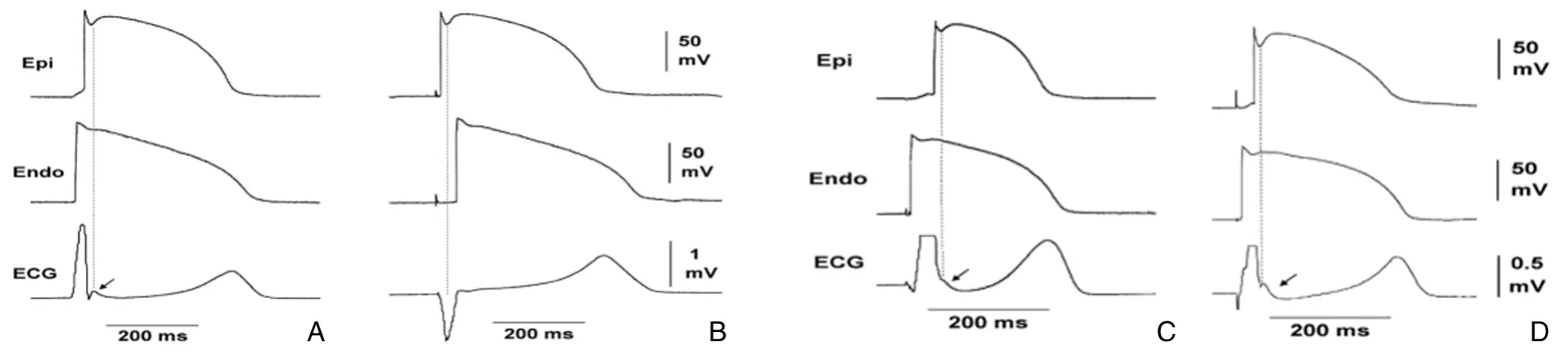
图1 J波的产生
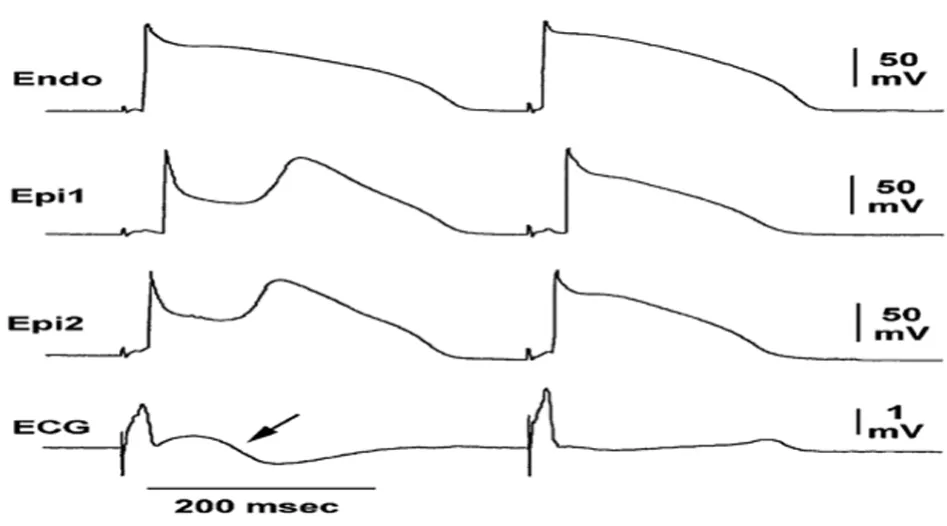
图2 心外膜动作电位切记加深,圆顶延迟出现,心电图表现出下斜形ST段抬高(箭头处)以及动作电位时程延长;Epi:心外膜;Endo:心内膜;ECG:心电图
(1)SCN5A-Nav1.5α亚基:SCN5A基因编码心脏钠离子通道 Nav1.5的 α亚基孔道,其突变与BrS1相关[16]。SCN5A位于染色体3p21位点,突变达100多种。与BrS1相关突变多属于功能缺失性突变,通过多种机制导致INa电流减弱,包括电压与时间依赖性的INa通道在激活、失活以及再激活之间转换的相关基因表达减弱;INa通道进入失活的中间状态后复苏减慢;加速INa通道失活[17]。G351V就是与BrS1相关一个典型的突变例子,其突变结果使INa电流衰减加速,从而减弱了 INa电流的密度[18]。Nav1.5具有3个门控过程,分别为激活、快速失活、缓慢激活。因此,BrS1的电生理表型取决于影响的门控过程以及程度[19]。实验证明,SCN5A基因突变的电生理表型具有温度依赖性,这与儿童发热后出现 BrS的表现相一致[20]。(2)GPD1L:GPD1L基因编码3磷酸甘油脱氢酶-1相关蛋白,其突变与BrS2相关。GPD1L位于染色体3p22-p24位点,主要表达于心肌细胞。GPD1L基因上的A280V突变可能通过减少钠通道到细胞膜的转运而影响SCN5A表面膜蛋白密度以及功能通道数量,但具体机制不清[21]。(3)SCN1B-Nav1.5β1亚基:SCN1B基因编码钠离子通道Nav1.5β1和β1b辅助亚基,起调节INa电流强度的作用。SCN1B基因位于染色体19q13.1位点,其突变与 BrS5相关[22]。β1和β1b蛋白由N端细胞外功能域(此为跨膜功能域,类似免疫球蛋白功能域)和C端细胞内功能域两部分组成[23]。SCN1B基因有3种突变亚基:p.Glu87 Glnβ1、p.Glu87Glnβ1B、 p.Trp179X β1B。 p.Trp179Xβ1B突变亚基蛋白缺乏细胞内和细胞外功能域,不能与肌膜和Nav1.5通道发生作用,使INa电流减弱,但Nav1.5通道激活与失活的电压依赖性却与野生型相似。另一种突变亚基p.Glu87Gln,具有细胞内功能域,能调控Nav1.5通道的门控特性(电压依赖性失活的转变),产生明显的负性作用。进一步实验证明,β1和β1b辅助亚基对Nav1.5通道正常调控取决于细胞外功能域[24]。综上所述,由于突变亚基缺失细胞外功能域,不能对Nav1.5通道发挥调节作用,导致 INa减弱。(4)SCN3B-Nav1.5β3亚基:SCN3B基因编码钠离子通道Nav1.5β3亚基,其结构和功能与β1相似,基因突变也引起INa电流减弱。SCN3B基因位于染色体 11q23位点[25],L10P为发生于SCN3B上一段高度保守残基序列的错义突变。此突变减弱β3亚基与Nav1.5通道相互作用以及Nav1.5到细胞膜的转运,从而影响钠离子通道功能及在细胞膜上表达,使INa电流减弱[26]。(5)CACNB2-Cav1.2β2亚基:CACNB2基因编码 L型电压门控钙通道(Cav1.2)的β2亚基,其突变与BrS4相关[27]。β2亚基调节 Cav1.2通道门控,增强ICa电流[28]。CACNB2基因位于染色体10p12位点,其发生的错义突变S481L,干扰β2亚基对Cav1.2通道的刺激作用,引起 ICa电流减弱[19]。6)CACNA1C-Cav1.2α1亚基:CACNA1C基因编码 L型电压门控钙通道(Cav1.2)的α亚基孔道,其突变与 BrS3相关[29]。CACNA1C基因位于染色体12p13.33位点,G490R和 A39V为发生于CACNA1C上的两个错义突变,突变干扰 Cav1.2通道β亚基的结合,从而使去极化内向ICa电流减弱[19]。(7)KCNE3—MiRP2多肽:KCNE3基因编码氨基酸多肽MiRP2—电压门控钾通道的5个β亚基之一,其突变与BrS6相关[30]。KCNE3基因位于染色体11q13-q14位点,R99H是发生于KCNE3上的错义突变。KCNE多肽是一种跨膜多肽,其跨膜功能域是调节钾离子通道的关键结构。实验证明,当KCNE多肽与Kv4.3相互作用时,Ik幅度减小,说明KCNE多肽对心脏Kv4.3(心室负责Ito电流的主要通道)通道起抑制作用;而R99H突变转染后,Ik电流幅度增强,说明R99H突变解除了这种抑制作用,对 Kv4.3通道产生了正向效应[31]。因此,R99H获得功能性突变使Ik增强引起了BrS6表型的产生。(8)KCNJ8—Kir6.1亚基:KCNJ8基因编码 ATP敏感性钾通道(IK-ATP)孔道亚基—Kir6.1,S422L为发生在KCNJ8上的错义突变,与ERS以及BrS相关。IK-ATP通道活性受细胞内ATP浓度的调控,正常ATP浓度下,仅有极少量 IK-ATP通道处于开放状态,实验证明开放率达到1.2%足以导致动作电位缩短50%[32]。IK-ATP通道的结构具有不均一性,其中Kir6.1主要在心外膜表达。S422L突变是一种功能获得性突变,由于突变导致IK-ATP通道对ATP敏感性降低,在正常ATP浓度下开放率增加,IK电流增大[33]。因此,Kir6.1在心肌的表达特点以及功能获得性突变构成了ERS的发生基础。
三、获得性J波综合征
J波综合征除了先天性基因突变以外,也具有后天获得性。后天获得性J波综合征与遗传性J波综合征具有相似的特性,包括心电图表现与室颤机制。最早发现低体温可诱导心电图出现明显J波以及室颤发生。当体温低于32℃时,心电图可出现J波,这是由于Ito和ICa通道的激活温度系数不同引起的。低体温状态下,ICa通道激活变缓慢,与Ito通道的对抗作用就减弱,使动作电位1相复极化程度增强,心外膜动作电位切记加深,于是心电图出现明显的J波。进一步发展出现动作电位圆顶消失,2相折返,可引发室颤的机制与J波综合征的电生理机制是相同的[34]。
另一个更常见类型的获得性J波综合征是心肌缺血诱发J波综合征。实验证明急性心肌缺血可引起Ito介导的心外膜动作电位圆顶消失,其诱发心电图ST段抬高的机制在某种程度上与J波综合征是相似的。更重要的是Ito介导的心外膜动作电位圆顶消失的不均一性促进了2相折返机制的发生,从而在心电图上表现出R-on-T偶联的室性期前收缩以及室颤[35]。当突发冠脉供血不足时,心肌细胞缺氧,代谢减弱,能量供应不足,离子通道功能降低,导致内向电流INa和 ICa减弱,而外向电流IK-ATP增强,从而打破了正常动作电位1相和2相的电流平衡,引发心律失常[36]。另一方面,局部性缺血使缺血区与非缺血区形成了缺血性边界,Ito介导的心外膜动作电位圆顶消失的不均一性在缺血性边界部位更加严重,促进了2相折返,产生局部再兴奋。紧邻缺血性边界发生的2相折返以及透壁性扩散导致了心电图R-on-T偶联性室性期前收缩以及室颤的发生[37]。
四、总结
经过多年的研究,对J波综合征相关发病机制的认识取得了重大进展,同时也揭示了与室颤等致死性心律失常的关系,促进了临床对心脏性猝死的预防与治疗,降低了死亡率。但在其机制与早期诊断方面仍然存在疑惑,需要进一步的研究来解决。
[1]BadriM,Patel A,Yan GX.Cellular and ionic basis of J-wave syndromes[J].Trends Cardiovasc Med,2015,25(1):12-21.
[2]Antzelevitch C,Yan GX.Jwave syndromes[J].Heart Rhythm,2010,7(4):549-558.
[3]Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic Jwave[J].Circulation,1996,93(2):372-379.
[4]Niwa N,Nerbonne JM.Molecular determ INants of cardiac transient outward potassium current(I(to))expression and regulation[J].JMol Cell Cardiol,2010,48(1):12-25.
[5]Wettwer E,Amos G J,Posival H,et al.Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin[J].Circ Res,1994,75(3):473-482.
[6]Nabauer M,Beuckelmann D J,Uberfuhr P,et al.Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle[J].Circulation,1996,93(1):168-177.
[7]Fish JM,Antzelevitch C.Role of sodium and calcium channel block in unmasking the Brugada syndrome[J].Heart Rhythm,2004,1(2):210-217.
[8]Antzelevitch C.Genetic,molecular and cellular mechanisms underlying the J wave syndromes[J].Circ J,2012,76(5):1054-1065.
[9]BadriM,Patel A,Yan GX.Cellular and ionic basis of J-wave syndromes[J].Trends Cardiovasc Med,2015,25(1):12-21.
[10]Antzelevitch CJ wave syndromes: molecular and cellular mechanisms[J].JElectrocardiol,2013,46(6):510-518.
[11]Yan G X,Antzelevitch C.Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation[J].Circulation,1999,100(15):1660-1666.
[12]Yan GX, Lankipalli RS, Burke JF, et al. Ventricular repolarization components on the electrocardiogram:cellular basis and clinical significance[J].JAm Coll Cardiol,2003,42(3):401-409.
[13]Bloch TP,Joergensen RM,Kanters JK,etal.Phase2 reentry in man[J].Heart Rhythm,2005,2(8):797-803.
[14]RuDusky BM.Rightbundle branch block,persistent ST-segment elevation,and sudden death[J].Am JCardiol,1998,82(3):407-408.
[15]Sethi KK,Sethi K,Chutani SK.Early repolarisation and Jwave syndromes[J].Indian Heart J,2014,66(4):443-452.
[16]Gellens ME,George AJ,Chen LQ,et al.Primary structure and functional expression of the human cardiac tetrodotoxininsensitive voltage-dependent sodium channel[J].Proc Natl Acad Sci U SA,1992,89(2):554-558.
[17]Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome:report of the second consensus conference[J].Heart Rhythm,2005,2(4):429-440.
[18]Vatta M,Dumaine R,Antzelevitch C,et al.Novelmutations in domain I of SCN5A cause Brugada syndrome[J].Mol Genet Metab,2002,75(4):317-324.
[19]Hedley P L,Jorgensen P,Schlamowitz S,et al.The genetic basis of Brugada syndrome:amutation update[J].Hum Mutat,2009,30(9):1256-1266.
[20]Rivolta I,Abriel H,Tateyama M,et al.Inherited Brugada and long QT-3 syndromemutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes[J].JBiol Chem,2001,276(33):30623-30630.
[21]London B,Michalec M,MehdiH,etal.Mutation in glycerol-3-phosphate dehydrogenase 1 like gene(GPD1-L)decreases cardiac Na+current and causes inherited arrhythmias[J].Circulation,2007,116(20):2260-2268.
[22]Makita N,Sloan-Brown K,Weghuis D O,et al.Genomic organization and chromosomal assignment of the human voltagegated Na+channel beta 1 subunit gene(SCN1B)[J].Genomics,1994,23(3):628-634.
[23]Qin N,D'Andrea M R,Lubin M L,etal.Molecular cloning and functional expression of the human sodium channel beta1B subunit,a novel splicing variant of the beta1 subunit[J].Eur J Biochem,2003,270(23):4762-4770.
[24]Watanabe H,Koopmann T T,Le Scouarnec S,et al.Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans[J].JClin Invest,2008,118(6):2260-2268.
[25]Morgan K,Stevens E B,Shah B,et al.beta 3:an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics[J].Proc Natl Acad Sci U SA,2000,97(5):2308-2313.
[26]Hu D,Barajas-Martinez H,Burashnikov E,etal.Amutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype[J].Circ Cardiovasc Genet,2009,2(3):270-278.
[27]Van Petegem F,Clark K A,Chatelain FC,etal.Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain[J].Nature,2004,429(6992):671-675.
[28]CatterallWA,Perez-Reyes E,Snutch T P,et al.International Union of Pharmacology.XLVIII.Nomenclature and structurefunction relationships of voltage-gated calcium channels[J].Pharmacol Rev,2005,57(4):411-425.
[29]Cardiocyte Cytoskeleton in Hypertrophied Myocardium[Z].
[30]Abbott GW,Butler M H,Bendahhou S,et al.MiRP2 forms potassium channels in skeletalmuscle with Kv3.4 and is associated with periodic paralysis[J].Cell,2001,104(2):217-231.
[31]Delpon E,Cordeiro JM,Nunez L,et al.Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome[J].Circ Arrhythm Electrophysiol,2008,1(3):209-218.
[32]Yokoshiki H,Sunagawa M,Seki T,et al.ATP-sensitive K+channels in pancreatic,cardiac,and vascular smooth muscle cells[J].Am JPhysiol,1998,274(1 Pt1):C25-C37.
[33]Medeiros-Domingo A,Tan B H,Crotti L,et al.Gain-offunction mutation S422L in the KCNJ8-encoded cardiac K(ATP)channel Kir6.1 as a pathogenic substrate for J-wave syndromes[J].Heart Rhythm,2010,7(10):1466-1471.
[34]Gurabi Z,Koncz I,Patocskai B,et al.Cellular mechanism underlying hypothermia-induced ventricular tachycardia/ventricular fibrillation in the setting of early repolarization and the protective effectof quinidine,cilostazol,andmilrinone[J].Circ Arrhythm Electrophysiol,2014,7(1):134-142.
[35]Yan GX,Joshi A,Guo D,et al.Phase 2 reentry as a trigger to initiate ventricular fibrillation during early acute myocardial ischemia[J].Circulation,2004,110(9):1036-1041.
[36]Carmeliet E.Cardiac ionic currents and acute ischemia:from channels to arrhythmias[J].Physiol Rev,1999,79(3):917-1017.
[37]Ehlert FA, Goldberger JJ. Cellular and pathophysiological mechanisms of ventricular arrhythmias in acute ischemia and infarction[J].Pacing Clin Electrophysiol,1997,20(4 Pt 1):966-975.
Research progress on mechanism s of J wave syndrome
Song Huanqiu,Liu Yuansheng.Emergency Department of Peking University People′s Hospital,Peking 100044,China
Jwave syndrome has emerged as a significant cause of idiopathic ventricular fibrillation(IVF)responsible for sudden cardiac death.Although J-wave syndrome involves a variety of types,but its mechanism and ECG performance are similar.The Jwave is the result of the 1-phase repolarization of the action potential,which ismainly generated by the Ito-mediated transient outward potassium flow.Because of the heterogeneity of Itodensity between the endocardium and epicardium,its heterogeneity constitutes the physiological basis of Jwave.In addition to Ito,other ion channels such as INa,Ica,Ikare also related to the generation of Jwaves.So any of anomalies of these ion channels can cause Jwaves appearing.The most common lesion of ion channel is gene mutation.A large number of experiments have shown that J wave syndrome is associated with a variety of ion channel genemutations such as SCN5A,CACNA1C,CACNB2,SCN1B,KCNE3 and so on.Themutation affects the epicardium repolarization,resulting in 2-phase reentry,triggering ventricular fibrillation.Therefore,a full understanding the pathomechanism of Jwave syndrome,contributing to clinical diagnosis and treatment,reducing themortality asmuch as possible.
J-wave syndrome; ERS; BrS; Ion channel; Genemutation
Correspondence author:Liu Yuansheng,Email:lyspku@126.com
10.3877/cma.j.issn.2095-6568.2017.02.001
刘元生,Email:lyspku@126.com
宋焕秋,刘元生.J波综合征发生机制的研究进展[J/CD].中华心脏与心律电子杂志,2017,5(2):65-69.
