肯尼迪病患者的临床表型与基因型分析
2017-10-17康健捷邓兵梅黎振声杨红军彭凯润王伟民
康健捷, 邓兵梅, 黎振声, 杨红军, 彭凯润, 王伟民
肯尼迪病患者的临床表型与基因型分析
康健捷1, 邓兵梅1, 黎振声2, 杨红军1, 彭凯润1, 王伟民3
目的探讨肯尼迪病(Kennedy’s disease,KD) 患者的临床特征和基因特点,以加强对KD的认识,减少误诊漏诊率。方法纳入2013年1月~2017年4月广州军区广州总医院神经内科收治的8例经基因确诊的KD患者,分析其临床特征、实验室检查、肌电图、神经电图和基因特点,使用肌萎缩侧索硬化症评分量表(amyotrophic lateral sclerosis rating scale,ALSFRS) 作为运动功能量表进行病情评估,分析临床特征及其与CAG重复序列数目的关系。结果所有患者均为成人发病,平均年龄为(36.63±4.14)岁,确诊病程平均为(12.13 ±3.44) y,均表现为四肢无力和肌肉萎缩,6例出现舌肌萎缩和构音障碍、肢体震颤、口周面肌束颤,4例性功能下降,6例乳腺发育。实验室检查结果7例肌酸激酶(creatine kinase,CK) 增高,8例甘油三酯增高,6例尿酸增高,2例睾酮增高。肌电图提示所有患者均表现为广泛神经源性损害,运动神经和感觉神经动作电位波幅降低,部分神经传导速度下降。AR基因CAG重复序列的重复次数为44~58次,CAG拷贝数与发病年龄呈负相关 (r=-0.753,P=0.031),与ALSFRS评分呈负相关 (r=-0.733,P=0.039),与CK 水平无关(r=0.250,P=0.550)。结论KD的临床特点为缓慢进展的延髓和脊髓肌肉萎缩无力,伴有肢体震颤、面肌束颤,部分可有内分泌功能及代谢紊乱。CAG 拷贝数越多,则发病年龄越早,运动功能评分越低。CAG拷贝数可作为KD病情的预测指标。
肯尼迪病; 基因诊断; 临床特点; 雄激素受体基因; 肌萎缩侧索硬化症评分量表
Abstract:ObjectiveTo strengthen the understanding of KD and avoid delayed diagnosis,we analyzed clinical,laboratory,electrophysiological and genetic characteristics of KD cases.MethodsEight cases diagnosed by gene as KD patients,were admitted for the study during January 2013 to April 2017.In this study,we analyzed the clinical manifestations,laboratory examination,EMG and genetic characteristics and used ALSFRS as motor function scores for condition assessment. The correlation between clinical features and CAG repeat size was analyzed.ResultsAverage age of onset was (36.63 ± 4.14) years and the average confirmed course were (12.13 ±3.44)years. Clinical features included medulla oblongata and spinal muscular atrophy and weakness in all eight cases,limbs tremor、perioral muscles twitch and gynecomastia in six cases,sexual dysfunction in four cases. Some patients had increased endocrine symptoms and metabolic disorders. EMG detected a widespread neuronal damage in all cases,and the sensory conductions were abnormal besides the motor conductions.The CAG repeat number in AR gene was from 44 to 58,respectively.The copy number of (CAG) n was negatively correlated with the onset age (r=-0.753,P=0.031) and the ALSFRS score (r=-0.733,P=0.039). The copy number of (CAG) n was independent of CK level(r=0.250,P=0.550).ConclusionKD is mainly presented with spinal and bulbar muscle atrophy and weakness,associated with endocrine and metabolic disturbance. The more copies,the early the onset and the lower the motor function score. The copy number of (CAG) n is valuable for assessment of KD.
Keywords: Kennedy’s disease; Clinical features; Gene analysis; Androgen receptor gene; Amyotrophic lateral sclerosis rating scale
肯尼迪病(Kennedy’s disease,KD)是一种晚发的X染色体连锁隐性遗传性神经系统变性疾病。该病于1968 年由 Kennedy 等[1]首次报道,一般成年早期起病,以慢性进展的肢体近端及球部肌无力肌萎缩为主要特征,同时合并有内分泌系统紊乱[2]。其发病率为 0.09/10 万,患病率为1.6/10 万,男性人群患病率约为 1/5万[3],临床对此病国外报道较多,国内报道较少。KD 临床表现常不典型,极易被误诊,从发病到确诊时间较长。现总结8例KD患者,结合文献复习,探讨该病临床特征、实验室检查、神经电生理特点和基因特点,对KD患者的病情进行定量评价,并分析临床特征与CAG重复序列数目的关系,以期有助于临床医生提高早期诊断率、掌握病情的发展程度和判断预后。
1 资料与方法
1.1 资料收集 纳入2013年1月~2017年4月广州军区广州总医院神经内科收治的8例KD患者,均以基因检测确诊。
1.2 研究方法
1.2.1 实验室检查 所有患者行血、尿常规、肝肾功能、血糖、电解质、心肌酶谱、凝血四项、糖化血红蛋白、癌胚抗原、甲胎蛋白、甲状腺功能全套、睾酮、催乳素、促卵泡生成素、雌二醇等生化指标测定。
1.2.2 神经电生理检查 所有患者应用肌电图仪进行常规肌电图及神经传导测定。对胸锁乳突肌、舌肌、肱二头肌、第一骨间肌、拇短展肌、T11椎旁肌、股四头肌、腓肠肌、胫前肌行肌电图检测。对双侧正中神经、尺神经、腓总神经、胫神经行运动和感觉神经行神经传导测定。所有患者行四肢体感诱发电位检查。测定结果均与本院电生理检查室正常值进行比较和判断。
1.2.3 雄激素受体(androgen receptor,AR)基因检测 经患者知情同意后,采集患者外周静脉血2 ml,送往广州市金域检验中心进行AR基因检测。
1.2.4 病情评估 使用肌萎缩侧索硬化症评分量表(amyotrophic lateral sclerosis rating scale,ALSFRS) 作为运动功能量表[4],进行评分,包括语言、流涎、吞咽、书写、切分食物及手握持器皿、穿衣和个人卫生、卧床和调整被褥、行走、上楼梯、呼吸10 个条目,条目采用0~4分5级评分法,满分40分为正常,功能完全丧失记0分。

2 结 果
2.1 临床特点
2.1.1 一般资料 8例 KD患者均为男性,年龄41~59岁,平均(48.75±6.09)岁,发病年龄31~48岁,平均(36.63±4.14)岁,从发病到基因确诊平均为(12.13±3.44) y。患者1曾被误诊为进行性肌营养不良,患者4曾被误诊为线粒体肌病,患者5曾被误诊为多发性肌炎,患者6曾被误诊为重症肌无力,其余4例患者曾被误诊为运动神经元病。8例患者均无阳性家族史。
2.1.2 临床症状和体格检查 8例患者临床症状(见表 1)均表现为隐匿起病,缓慢进展。首发症状:下肢乏力4例,四肢乏力2例,咀嚼乏力1例,性功能下降1例。8例均有肢体乏力,以近端受累为主,表现为上楼、跑步困难,下蹲后站起费力,双臂抬举费力,提重物困难。6例舌肌萎缩(见图 1)、吐字欠清、口周肌肉跳动、肢体肌肉萎缩和上肢发抖,2例有饮水呛咳,6例经超声波诊断有乳腺发育(见图 1),4例性功能下降,1例弱精症(患者5)。
体格检查见表 1,可见6例患者舌肌萎缩和震颤、构音障碍、口周肌肉束颤、肢体肌肉萎缩和双上肢震颤,8例患者四肢近端肌力 3~5 级,远端肌力4~5级,5例腱反射消失,3例腱反射减弱,所有患者均未引出病理征,深浅感觉检查正常,无认知功能下降。
2.2 实验室检查 见表2,7例CK升高(476~2306 IU/L);2例血清性激素异常,睾酮增高,8例血脂异常,其中4例甘油三酯及低密度脂蛋白升高,另外4例单独甘油三酯升高;6例尿酸升高;血、尿常规、肝肾功能、血糖、电解质、凝血四项、糖化血红蛋白、癌胚抗原、甲胎蛋白、甲状腺功能全套均正常。
2.3 电生理检查 肌电图显示8例患者均表现为广泛的神经源性损害。神经传导检查显示6例患者感觉神经和运动神经动作电位波幅均降低,潜伏期正常,2例传导速度下降,其中患者1、患者2和患者7的胫神经和腓总神经感觉动作电位未引出。1例(患者4)感觉神经动作电位波幅均为正常低限值,运动神经传导正常。1例(患者6)感觉神经动作电位波幅降低,运动神经传导正常。除了患者4,其余7例患者四肢体感诱发电位均异常,提示上下肢周围神经受累。
2.4 基因检测 8例患者的AR基因CAG三核苷酸的重复序列数均超过40(见表1)。患者2的 AR基因CAG三核苷酸的重复序列数为53次(见图2)。
2.5 运动功能评分 8例患者的 ALSFRS量表评分见表1。
2.6 患者CAG拷贝数 与发病年龄呈负相关 (r=-0.753,P=0.031),与ALSFRS评分呈负相关 (r=-0.733,P=0.039),提示CAG 拷贝数越多,则发病年龄越早,ALSFRS评分越低,病情可能越严重。病程与ALSFRS评分呈负相关趋势(r=-0.434,P=0.283),提示尚无统计学意义。患者CAG拷贝数与CK 水平无关(r=0.250,P=0.550)(见图3)。
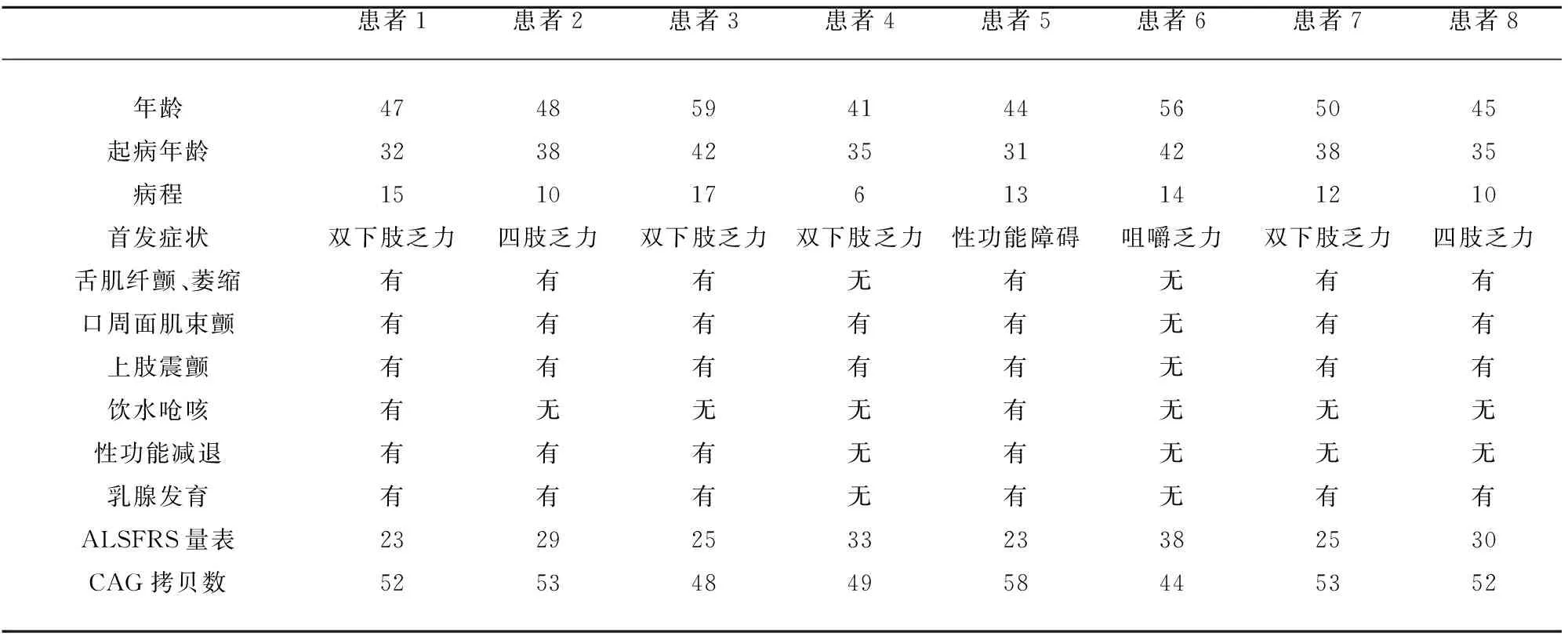
表1 8例患者的一般资料和临床症状
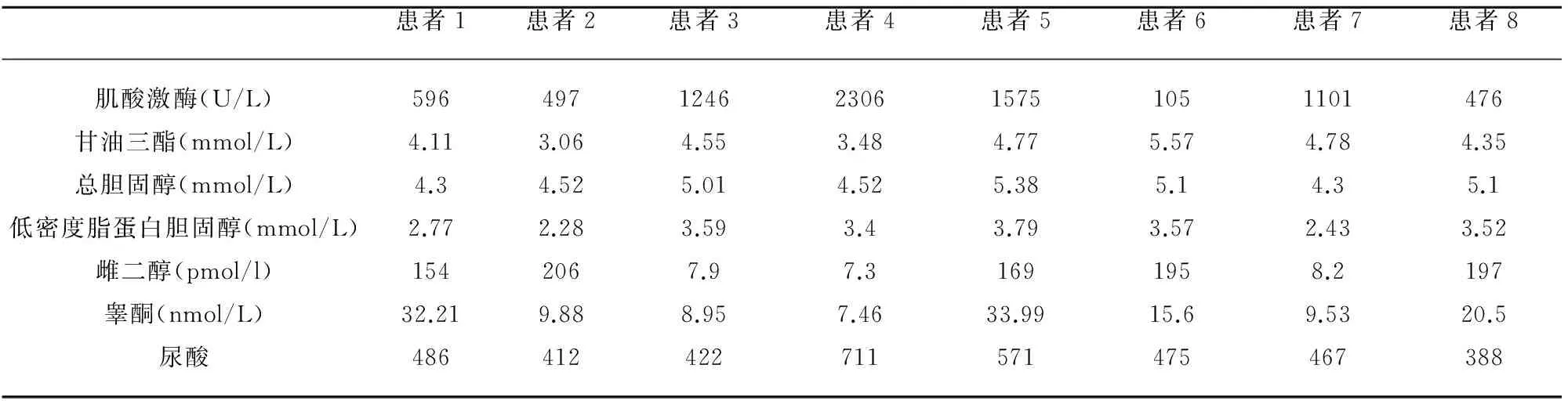
表2 8例患者的实验室检查结果
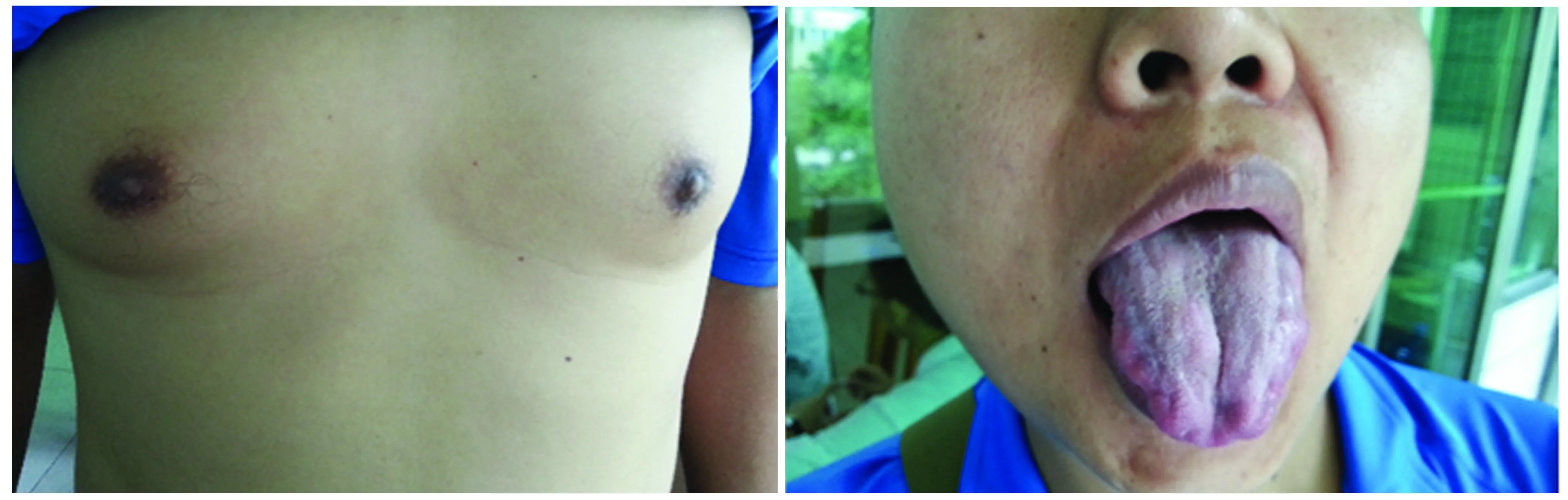
图1 患者2乳房女性化发育和舌肌萎缩
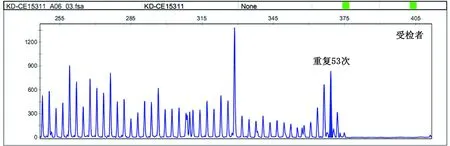
图2 患者2的AR基因CAG三核苷酸的重复序列数为53次
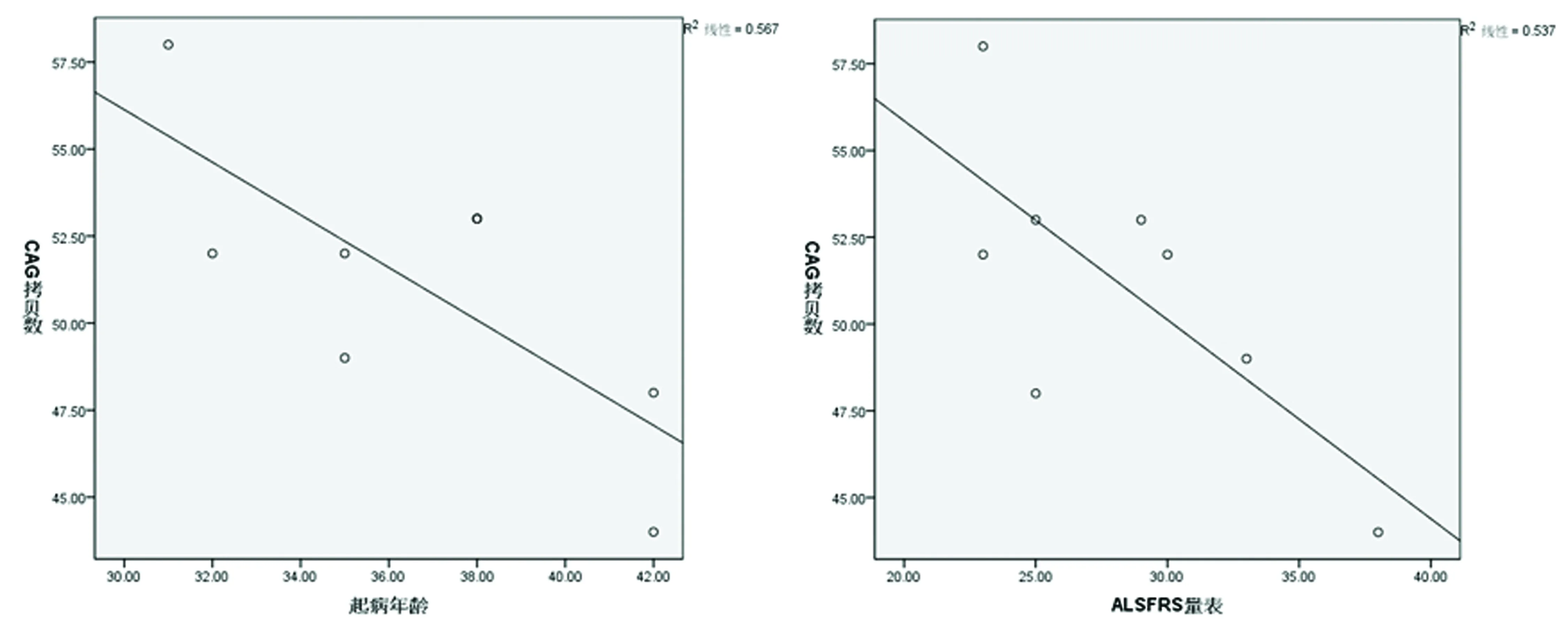
图3 CAG拷贝数与起病年龄、ALSFRS评分的相关性散点图
3 讨 论
KD又称脊髓延髓肌肉萎缩症(spinal and bulbar muscular atrophy,SBMA),主要累及下运动神经元、感觉神经和内分泌系统,临床较罕见,诊断主要依据患者的临床症状、CK 水平、肌肉病理活检及神经电生理结果,最终需进行雄激素受体基因第1个外显子CAG重复序列异常扩增次数以确诊[2]。在正常个体,雄激素受体基因第一个外显CAG基因重复数一般在11~35次,而在患者和女性携带者中该等位基因的CAG基因重复数一般在40~62次。本研究中8例患者的CAG三核苷酸的重复序列数均超过40,且发现CAG拷贝数与KD起病年龄呈负相关,与ALSFRS评分呈负相关,推测CAG 拷贝数越多,则发病年龄越早,临床运动功能损害越严重,预后相对较差。
文献报道[5~7],KD属于晚发疾病,发病年龄多在20~50岁,进展缓慢,常从下肢近端渐进性肌肉无力开始,逐渐波及到下肢远端及上肢,多伴有肌肉萎缩,常出现肌束颤动,以面肌显著,舌肌、咀嚼肌以及延髓肌亦常被累及,从而引起吐字不清,而吞咽困难及饮水呛咳出现相对较晚,常合并雄激素不敏感症状,如乳房增大,睾丸缩小以及性能力低下,还可出现姿势性震颤。本研究中8例 KD 患者的临床表现与典型KD相符合,均为中年起病,病情缓慢进展,有延髓和脊髓下运动神经元受损以及乳腺发育。其中6例以肢体乏力起病,另2例分别以咀嚼乏力和性功能下降起病,2~3 y后也出现四肢乏力。患者肢体无力均以近端为主,手部精细活动保存完好。6例患者有舌肌萎缩、舌肌震颤和构音障碍,但只有2例患者有饮水呛咳,5例患者病程超过10 y仍尚未出现吞咽功能障碍。实验室检查显示7例CK升高,提示肌肉系统受到损害。2例睾酮增高,提示雄激素功能不正常。6例尿酸增高,7例血脂异常,提示代谢紊乱,而空腹血糖、甲状腺功能均未见异常,与既往其他报道相一致[7~9]。6例患者出现内分泌异常的症状和体征,包括乳腺发育(6例) 和性功能障碍(4例),乳腺发育病例的比例(6/8)较以往报道偏高[7~9],推测原因可能是本研究中每例患者均通过超声诊断是否存在乳腺发育,所以减少了因为单纯的触诊而漏诊。患者5的CK正常,患者1、患者2和患者8的CK轻度升高,其CAG 拷贝数分别达到了52、53、52,而患者4的CAG 拷贝数是49,CK却显著升高,高达2306U/L,病程6 y,临床肢体乏力症状很轻。通过数据分析得知,患者CK 水平与肌力改变、病程以及ALSFRS评分均无关,CAG拷贝数与CK 水平也无关,提示患者的肌无力和运动功能下降并非主要由肌肉系统损害所致,CK不能作为判断病情和预后的因素。研究证实[10,11],CAG重复次数异常增多造成AR蛋白氨基端的多谷氨酰胺链延长,这种突变的雄激素受体与配体睾酮结合后即由胞浆转入细胞核内,其毒性作用和降解异常等机制最终导致细胞变性坏死,而AR蛋白在脊髓和脑干运动神经元、初级和次级生殖器官、非生殖器官和骨骼肌均有表达,因此,临床出现神经、肌肉损害及男性特征减退。推测引起肌酶升高的原因可能是肌纤维直接被破坏和失神经支配引起肌肉萎缩导致肌纤维再生障碍所致,临床表现的舌肌萎缩和肢体无力主要由脑干和脊髓运动神经元损害所致。
本研究中肌电图显示8例患者均表现为广泛的神经源性损害。6例感觉神经动作电位波幅和运动神经动作电位波幅均降低,3例的胫神经和腓总神经感觉动作电位甚至未引出,提示感觉神经损害严重。但是,临床上患者无肢体麻木主诉,查体也未见痛温觉下降,所以感觉系统损害极易被忽视,从而易被误诊为只累及运动系统的运动神经元病。因此,在鉴别KD和运动神经元病时,电生理检查结果要特别引起注意,尤其是感觉神经动作电位波幅有无降低或有无引出。另外,患者4感觉神经动作电位波幅均在正常低限,而运动神经动作电位均正常,患者6感觉神经动作电位波幅明显降低,而运动神经动作电位均正常,提示感觉神经损害可能早于并重于运动神经。
在临床上,目前常用ALSFRS量表对KD患者进行临床评估。神经功能缺损越严重则得分越低。本研究使用 ALSFRS 量表进行评分发现,KD 患者失分的项目主要集中在肢体运动功能下降,如书写、穿衣和个人卫生、行走、上楼梯等方面,而言语功能及吞咽功能下降不明显。本研究发现CAG 拷贝数越多,则发病年龄越早,ALSFRS评分越低,病情可能越严重。提示CAG拷贝数可作为KD病情的预测指标。而病程与ALSFRS评分呈负相关趋势,但是尚无统计学意义,考虑可能是研究样本量偏少所致。
目前,KD尚缺乏特异性治疗方法,降低男性患者的雄激素可用于治疗KD,“亮丙瑞林” 对于病程<10 y的患者,可延缓病情进展,但疗效有限[12]。随着对KD发病机制研究的不断深入,一些新的药物临床研究正在进行中。正因为缺乏特异性治疗方法,所以阻断KD在家系中的遗传非常重要。但是,因为KD与其他神经系统变性疾病的临床表现相似,所以极容易被临床忽略和误诊。目前病例报告的KD患者从首次出现症状到基因确诊的时间较长,本研究结果为(12.13±3.44) y,所以KD患者被确诊时往往其女儿已经到了生育年龄,而KD是一种X染色体连锁隐性遗传性疾病,所以,患者的女儿均为致病基因携带者,若在生育之前其父亲能被确诊,则这些女性携带者在怀孕时可进行产前胎儿基因诊断,以确保不携带致病基因的胎儿出生,从而阻断KD在家系中的遗传,因此,KD患者的早期确诊对患者家族的优生优育有重大意义。
综上所述,提高对KD的认识并尽早确诊十分重要,对于肢体无力伴肌肉萎缩进展缓慢的、舌肌萎缩病程长而无吞咽障碍的、无感觉障碍主诉而电生理检查提示感觉神经受损的、不明原因肌酸激酶升高的中老年男性,推荐乳房超声检查、性激素、血脂、尿酸检测,仔细询问生育史及性功能是否有减退,并尽早进行AR基因的CAG拷贝数检测,从而避免肌肉活检、腰椎穿刺术等有创检查,尽早明确诊断。总之,分析 KD 的临床特征、实验室检查、神经电生理和基因特点,寻找评价病情的发展程度和预后的相关因素,对临床医生减少误诊及延误诊断、掌握病情的发展程度和判断预后有帮助,对开展药物临床研究意义重大。
[1]Kennedy WR,Alter M,Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset:a sex-linked recessive trait[J]. Neurology,1968,18(7):671-680.
[2]Finsterer J. Perspectives of Kennedy’s disease [J]. J Neurol Sci,2010,298:1-10.
[3]La Spada AR,Wilson EM,Lubahn DB,et al. Androgen receptor genemutations in X-linked spinal and bulbar muscular atrophy[J]. Nature,1991,352:77-79.
[4]Kollewe K,Mauss U,Krampfl K,et al. ALSFRS-R score and its ratio:a useful predictor for ALS-progression[J]. J Neurol Sci,2008,275(1/2):69-73.
[5]Rhodes LE,Freeman BK,Auh S,et al. Clinical features of spinal and bulbar muscular atrophy[J]. Brain,2009,132(12):3242-3251.
[6]Grunseich C,Rinaldi C,Fischbeck KH. Spinal and bulbar muscular atrophy:pathogenesis and clinical management [J]. Oral Dis,2014,20(1):6-9.
[7]鲁 明,樊东升,李小英,等. 基因确诊的肯尼迪病两例临床与分子生物学特点[J]. 中华神经科杂志,2007,40(4):232-236.
[8]谢曼清,李晓光,崔丽英,等. 肯尼迪病基因诊断及临床特点[J]. 中华医学杂志,2010,90(35):2498-2500.
[9]鲁 明,樊东升,张 俊,等. 肯尼迪病患者27例临床特征[J]. 中华神经科杂志,2008,41(7):452-454.
[10]Adachi H,Waza M,Katsuno M,et al. Pathogenesis and molecular targeted therapy of spinal and bulbar muscular atrophy[J]. Neuropathol Applied Neurobiol,2007,33(2):135-151.
[11]Fratta P,Niranjanan N,Masset L,et al. Correlation of clinical and molecular features in spinal bulbar muscular atrophy[J]. Neurology,2014,82(23):2077-2084.
[12]Katsuno M,Banno H,Suzuki K,et al. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study):a multicentre,randomised,double-blind,placebo-controlled trial[J]. Lancet Neurol,2010,9(9):875-884.
TheclinicalcharacteristicsandgenemutationreportsofKennedy’sdisease
KANGJianjie,DENGBingmei,LIZhensheng,etal.
(DepartmentofNeurology,GeneralHospitalofGuangzhouMilitaryCommandofPLA,Guangdong510010,China)
1003-2754(2017)09-0813-05
2017-07-10;
2017-08-18
广东省医学科学技术研究基金项目(No. A2015084)
(1.广州军区广州总医院神经内科,广东 广州 510010;2.广州军区广州总医院癫痫科,广东 广州 510010;3.广州军区广州总医院神经外科,广东 广州 510010)
邓兵梅,E-mail:bingmeid@163.com
R746
A
