尼达尼布的合成新方法
2017-09-16赵菲熠邵玲艳冀亚飞
赵菲熠, 邵玲艳, 郭 瑛, 冀亚飞
(华东理工大学 药学院,上海 200237)
·制药技术·
尼达尼布的合成新方法
赵菲熠, 邵玲艳, 郭 瑛*, 冀亚飞*
(华东理工大学 药学院,上海 200237)
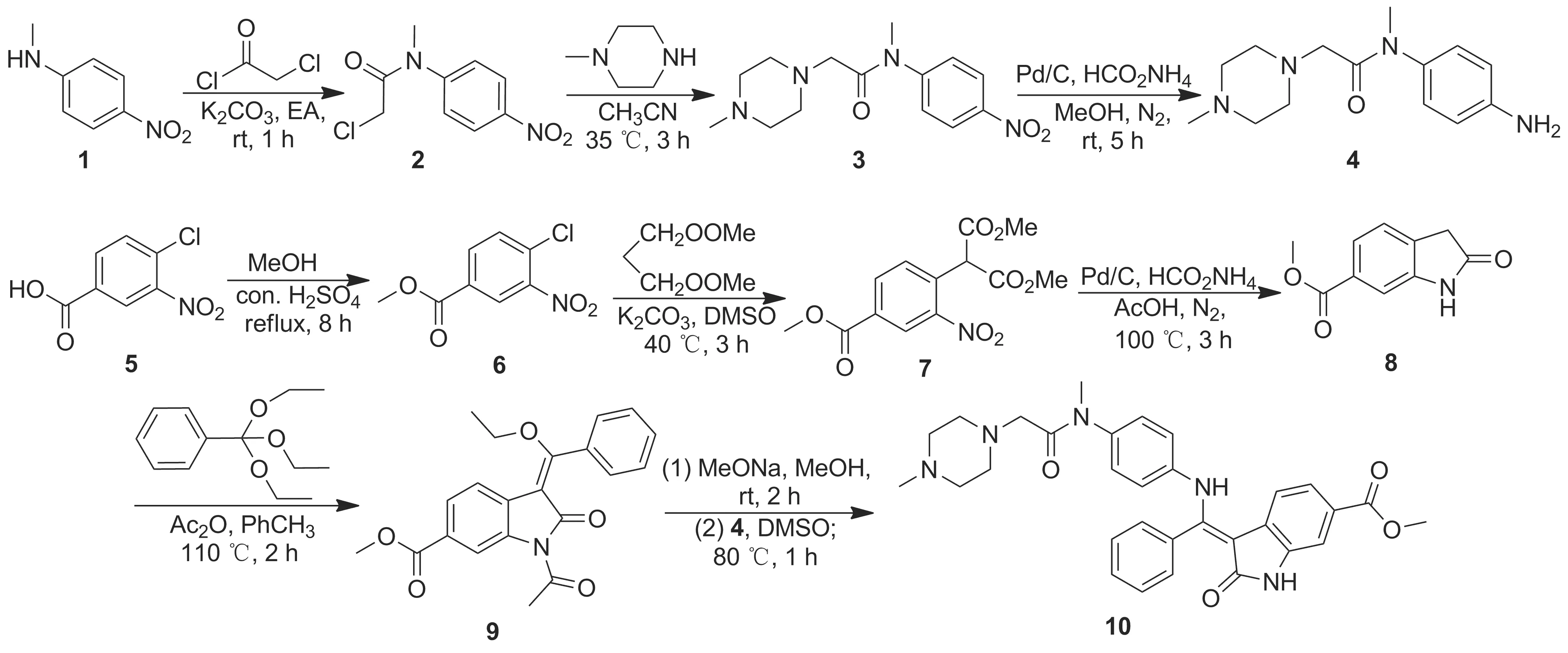
以N-甲基-4-硝基苯胺作为起始原料,依次经氯乙酰化、取代及氢化还原反应制得关键中间体N-(4-氨基苯基)-甲基-2-(4-甲基-1-哌嗪基)乙酰胺(4);以4-氯-3-硝基苯甲酸为原料,依次经酯化、取代、氢化还原及环合反应制得6-甲氧羰基-2-吲哚酮(8);8与原苯甲酸三乙酯和乙酸酐经“一锅煮”反应制得中间体1-乙酰基-3-甲氧基(苯基)亚甲烯基-2-氧代吲哚环-6-羧酸甲酯(9);4和9进行取代反应的同时脱除保护,经“一锅煮”反应合成尼达尼布,总收率57.2%,其结构经1H NMR,13C NMR和MS(ESI)确证。
N-甲基-4-硝基苯胺; 4-氯-3-硝基苯甲酸; 尼达尼布; 特发性肺纤维化; 酪氨酸激酶抑制剂; 一锅煮; 药物合成
尼达尼布(Nintedanib,10),化学名为(3Z)-2, 3-二氢-3-【【【4-{甲基[2-(4-甲基-1-哌嗪基)乙酰基]氨基}苯基】氨基】苯亚甲基】-2-氧代-1H-吲哚-6-甲酸甲酯,是勃林格殷格翰公司特别针对特发性肺纤维化(IPF)而开发的小分子酪氨酸激酶抑制剂(TKI)[1],也是首个获得临床证据一致证实、可显著减少肺功能年下降率(减少幅度可达50%),从而延缓IPF疾病进展的、IPF靶向治疗的药物[2-4],可同时阻断血小板源性生长因子受体(PDGFRα,β)、血管内皮生长因子受体(VEGFR1~3)和成纤维细胞生长因子受体(FGFR 1~3)。10的研制给IPF患者带来了希望,被美国食品药品管理局(FDA)授予突破性治疗药物的地位[5]。
Scheme1
目前文献公开报道的10的最新合成路线[6]是在前人报道的工艺路线[7-10]上的改进:由N-甲基4-硝基苯胺(1)为起始原料,经氯乙酰化、取代、氢化还原制得第一个关键中间体N-(4-氨基苯基)-N-甲基-2-(4-甲基-1-哌嗪基)乙酰胺(4);再以3-硝基苯甲酸为起始原料,经酯化、取代、氢化还原、环合及缩合等反应合成另一中间体1-乙酰基-3-甲氧基(苯基)亚甲烯基-2-氧代吲哚环-6-羧酸甲酯(9);4与9经取代反应后水解脱除保护制得10。该合成路线主要存在的问题包括[11]:(1)3-硝基苯甲酸甲酯与氯乙酸甲酯反应时存在异构化,影响反应的选择性;(2)该系列反应后处理时多次采用柱层析方式,操作繁琐且浪费大量溶剂;(3)中间体6-甲氧羰基-2-吲哚酮(8)与原苯甲酸三乙酯缩合时,对吲哚环的取代率较低,直接影响最终收率。另外,有研究人员报道了形成吲哚环的其他方法[12],该反应以4-甲基-3-硝基苯甲酸为原料,经酯化、取代、氧化脱羧、氢化还原环合制得8。该方法存在以下问题:(1)在第二步取代反应中,我们未能有效地重现实验结果;(2)氧化脱羧时,芳环上的酯基易发生水解反应,增加副反应。
本文在文献报道的基础上,对尼达尼布的合成工艺进行改进:以1为起始原料,经氯乙酰化、取代及氢化还原反应制得第一个关键中间体4;以5为起始原料,依次经酯化、取代、氢化还原及环合反应制得吲哚酮衍生物8;8通过乙酰化和缩合“一锅煮”反应制得中间体9; 随后4与9进行取代反应的同时水解脱除保护基团合成目标产物尼达尼布(10, Scheme 1),总收率57.2%,其结构经1H NMR,13C NMR和MS(ESI)确证。该合成路线原料易得、反应条件温和、选择性较好,具有规模化应用潜力。
1 实验部分
1.1 仪器与试剂
XT4A型显微熔点仪(温度未校正);Bruker AVANCE 400型核磁共振仪(CDCl3和DMSO-d6为溶剂,TMS为内标);LC/TOP MS型和QSTAR Pulsar I LC/TOF MS型质谱仪,Agilent 1260液相色谱仪。
所用试剂均为化学纯或分析纯;无水反应所用溶剂经4Å分子筛脱水处理。
1.2 合成
(1)N-(4-硝基)-N-甲基-2-氯乙酰胺(2)的合成
在反应瓶中依次加入13.75 g(25 mmol)、碳酸钾6.90 g(20 mmol)和乙酸乙酯(EA)30 mL,于室温搅拌,在恒压滴液漏斗中加入氯乙酰氯4.24 g(2.81 mL, 37.5 mmol)和EA 10 mL,逐滴滴加至反应液,于室温反应1 h。加水(3×30 mL)洗涤,分液,有机层用无水硫酸镁干燥,过滤,滤液浓缩,所得固体用冰乙醇重结晶,干燥得淡黄色固体25.49 g,收率96.2%, m.p.116~117 ℃;1H NMRδ: 8.33(d,J=11.6 Hz, 2H, ArH), 7.48(d,J=11.6 Hz, 2H, ArH), 3.94(s, 2H, CH2), 3.39(s, 3H, CH3);13C NMRδ: 166.0, 148.4, 146.9, 127.7(2C), 125.3(2C), 41.3, 38.0; MS(ESI)m/z: 229.0{[M+H]+}。
(2)N-(4-硝基苯基)-N-甲基-2-(4-甲基-1-哌嗪基)乙酰胺(3)的合成
在反应瓶中依次加入24.56 g(20 mmol)、甲基哌嗪2.20 g(22 mmol)和乙腈20 mL,搅拌下于35 ℃反应3 h。过滤,滤液浓缩,残余物加入乙酸乙酯30 mL,依次用饱和食盐水(3×30 mL)洗涤、无水硫酸镁干燥,浓缩得黄色液体粗品35.64 g,直接用于下步反应;1H NMRδ: 8.29(d,J=8.8 Hz, 2H, ArH), 7.43(d,J=8.8 Hz, 2H, ArH), 3.35(s, 3H, CH3), 3.08(s, 2H, CH2), 2.60(s, 8H, CH2), 2.38(s, 3H, CH3);13C NMRδ: 169.2, 149.4, 146.1, 127.2(2C), 124.8(2C), 60.4, 54.8(2C), 53.0(2C), 45.9, 37.4; MS(ESI)m/z: 293.2{[M+H]+}。
(3)4的合成
在反应瓶中依次加入粗品3、 5% Pd/C 0.50 g、甲酸铵12.60 g(200 mmol)和无水甲醇30 mL,氮气保护,于室温反应5 h。过滤,滤液浓缩后加入二氯甲烷30 mL,析出甲酸铵,过滤,滤液依次用饱和食盐水(3×30 mL)洗涤,无水硫酸镁干燥,浓缩,残余物用混合溶剂[乙酸乙酯/石油醚(V/V=1/1)]重结晶得淡黄色固体44.58 g,两步总收率87.4%, m.p.147~148 ℃;1H NMRδ: 6.93(d,J=8.4 Hz, 2H, ArH), 6.66(d,J=8.4 Hz, 2H, ArH), 3.19(s, 3H, CH3), 2.90(s, 2H, CH2), 2.53(s, 8H, CH2), 2.30(s, 3H, CH3);13C NMRδ: 169.8, 146.3, 133.8, 128.2(2C), 115.6(2C), 59.2, 54.8(2C), 53.0(2C), 45.8, 37.5; MS(ESI)m/z: 263.2{[M+H]+}。
(4) 4-氯-3-硝基-苯甲酸甲酯(6)的合成
在反应瓶中依次加入54.03 g(20 mmol)和无水甲醇20 mL,搅拌下缓慢滴加浓硫酸2 mL,滴毕,回流反应8 h。冷却至室温,缓慢滴加饱和碳酸氢钠溶液至pH 7~8,过滤,滤饼用冰水(3×20 mL)淋洗,红外干燥得白色固体64.14 g,收率96.2 %, m. p.83~84 ℃;1H NMRδ: 8.52(d,J=1.6 Hz, 1H, ArH), 8.17(d,J=8.4 Hz, 1H, ArH), 7.65(d,J=8.4 Hz, 1H, ArH), 3.97(s, 3H, CH3);13C NMRδ: 164.2, 147.9, 133.6, 132.2, 131.7, 130.1, 126.6, 53.0; MS(ESI)m/z: 238.1{[M+Na]+}。
(5) (4-甲氧羰基-2-硝基苯基)丙二酸二甲酯(7)的合成
在反应瓶中依次加入碳酸钾2.76 g(20 mmol)、丙二酸二甲酯1.45 g(11 mmol)和无水N,N-二甲基亚砜(DMSO)15 mL,搅拌下于室温反应2 min,分批加入62.16 g(10 mmol),于40 ℃反应3 h。加入乙酸乙酯30 mL,依次用1 mol·L-1盐酸(30 mL)和饱和食盐水(2×30 mL)洗涤,无水硫酸镁干燥,浓缩后残余物用乙酸乙酯重结晶,红外干燥得淡黄色晶体72.79 g,收率89.7%, m.p.73~74 ℃;1H NMRδ: 8.69(s, 1H, ArH), 8.29(d,J=8.0 Hz, 1H, ArH), 7.63(d,J=8.0 Hz, 1H, ArH), 5.37(s, 1H, CH), 3.99(s, 3H, CH3), 3.82(s, 6H, CH3);13C NMRδ: 167.1(2C), 164.5, 148.8, 134.0, 132.1, 132.0, 131.6, 126.3, 54.1, 53.4(2C), 52.9; MS(ESI)m/z: 334.0{[M+Na]+}。
(6)8的合成
在反应瓶中依次加入71.56 g(5 mmol)、 5%Pd/C 0.15 g、甲酸铵3.15 g(50 mmol)和冰醋酸10 mL,氮气保护,于100 ℃反应3 h。趁热过滤除去Pd/C(冷却极有可能导致产物大量析出),滤液减压蒸馏,残余液缓慢滴加饱和碳酸氢钠溶液至无气泡溢出,持续搅拌1 h,过滤,固体用水(10 mL)淋洗,红外干燥得白色粉末80.945 g,收率99.0%;1H NMRδ: 8.42(s, 1H, NH), 7.76(d,J=8.0 Hz, 1H, ArH), 7.55(s, 1H, ArH), 7.30(d,J=8.0 Hz, 1H, ArH), 3.92(s, 3H, CH3), 3.60(s, 2H, CH2);13C NMRδ: 176.0, 166.1, 144.1, 131.7, 128.8, 124.4, 122.5, 108.9, 40.1, 35.9; MS(ESI)m/z: 190.9[M+]。
(7)9的合成
在反应瓶中依次加入80.76 g(4 mmol)、甲苯4 mL和乙酸酐2 mL,搅拌下滴加原苯甲酸三乙酯2.69 g(12 mmol),滴毕,回流(110 ℃)反应2 h。冷却至室温,浓缩,加入饱和碳酸氢钠溶液15 mL,搅拌30 min,用乙酸乙酯(2×20 mL)萃取,合并萃取液,依次用饱和碳酸氢钠溶液(2×30 mL)和饱和食盐水(30 mL)洗涤,无水硫酸镁干燥,浓缩,残余物用石油醚重结晶,红外干燥得类白色固体91.32 g,收率90.4%, m.p.187~188 ℃;1H NMRδ: 8.91(s, 1H, ArH), 8.05(d,J=8.0 Hz, 1H, ArH), 7.96(d,J=8.0 Hz, 1H, ArH), 7.58~7.56(m, 3H, ArH), 7.41~7.39(m, 2H, ArH), 4.03~3.97(m, 2H, CH2), 3.93(s, 3H, CH3), 2.57(s, 3H, CH3), 1.44(t,J=14.0 Hz, 3H, CH3);13C NMRδ: 171.4, 171.3, 167.3, 167.1, 136.5, 131.3, 130.6, 129.0(2C), 128.5, 128.3(2C), 126.4, 122.4, 116.5, 105.8, 67.1, 52.2, 27.0, 15.4; MS(ESI)m/z: 388.1{[M+Na]+}。
(8)10的合成
在反应瓶中依次加入90.73 g(2 mmol)、甲醇钠0.22 g(4 mmol)和甲醇8 mL,搅拌下于室温反应2 h。加入40.69 g(2.4 mmol)和二甲基亚砜4 mL,升温至80 ℃,反应1 h。减压蒸馏除去甲醇,加入乙酸乙酯 20 mL,依次用水(3×20 mL)洗涤,无水硫酸镁干燥,浓缩后用无水甲醇重结晶,红外干燥得黄色固体100.95 g,收率88.1%;1H NMRδ: 12.17(s, 1H, NH), 8.66(s, 1H, NH), 7.59~7.51(m, 4H, ArH), 7.44~7.42(m, 2H, ArH), 7.38(d,J=8.0 Hz, 1H, ArH), 6.97(d,J=8.4 Hz, 2H, ArH), 6.80(d,J=8.8 Hz, 2H, ArH), 5.99(d,J=8.0 Hz, 1H, ArH), 3.85(s, 3H, CH3), 3.17(s, 3H, CH3), 2.79(s, 8H, CH2), 2.25(s, 3H, CH3);13C NMRδ: 171.1, 169.4, 167.4, 158.3, 139.8, 138.0, 135.7, 132.3, 130.6, 129.6(2C), 129.1, 128.5(2C), 127.8(2C), 125.1, 123.9(2C), 122.7, 118.2, 110.4, 98.5, 59.5, 54.8(2C), 53.1(2C), 51.9, 45.8, 37.4; MS(ESI)m/z: 540.2{[M+H]+}。
在化合物2的合成中,以廉价的碳酸钾代替通常使用的4-二甲氨基吡啶、三乙胺等有机碱以及反应效果最佳但昂贵的碳酸锂,不仅有利于降低成本,而且与文献使用的三乙胺相比,反应放热平稳,无需冰浴条件,实验操作和后处理更加简便,有利于工业化生产;溶剂筛选表明,使用乙酸乙酯作为反应溶剂,液相收率达到98%,而且在该条件下反应, 0~40 ℃收率均较高。
在化合物3的合成过程中,溶剂筛选结果表明:甲苯、乙腈和四氢呋喃的效果均好于文献使用的丙酮,考虑到溶剂的毒性及沸点,最终选择乙腈作为溶剂。该反应同样需要在碱的催化下进行,我们对常用的碱甲醇钠、乙醇钠、叔丁醇锂、碳酸锂和碳酸钠进行了筛选,实验表明碳酸锂的催化效果最佳,但综合考虑成本等因素,我们选择了液相收率可达到97%的碳酸钾,并使反应温度从60 ℃降低至35 ℃。
在化合物4和8的合成中,我们改变原有的氢气催化,使用了Pd/C-甲酸铵的催化体系,使反应脱离了氢气,极大提高了安全系数;值得注意的是,由于前者的产物4溶于水,后处理利用甲酸铵及还原产物在二氯甲烷中的难溶性,先用二氯甲烷析出无机盐,过滤后溶于乙酸乙酯,用饱和食盐水洗涤,减少4的损失。
在化合物6的合成中,主要的副反应是6的酯键水解,脱下来的醇与氯离子反应生成4-甲氧基-3-硝基苯甲酸,无水条件可对该反应起到极大的抑制作用,因此反应过程中要注意无水操作。使用甲醇钠为傅酸剂时,甲醇钠易与6的氯原子发生副反应,综合考虑效能及成本问题,选用碳酸钾作为傅酸剂,并通过溶剂筛选使液相收率达到95%以上,反应温度从80 ℃降低至45 ℃。后处理采用先水洗,再酸洗,后水洗的方式优先除去4-甲氧基-3-硝基苯甲酸,再使用乙酸乙酯重结晶,收率极高。
在化合物9的合成探究中,我们发现直接使用乙酸酐作为反应溶剂,产物停留在1-乙酰基-2-氧代吲哚环-6-羧酸甲酯,不会继续反应。本文采用甲苯-乙酸酐作为混合溶剂,并探究了改变甲苯-乙酸酐的比例对目标化合物收率的影响,最终采用了甲苯/乙酸酐体积比为2 ∶1的混合溶剂。
对尼达尼布的合成工艺进行了改进,反应总收率大大提高。分别由N-甲基-4-硝基苯胺(1)和4-氯-3-硝基苯甲酸(5)为起始原料合成了尼达尼布的两个关键中间体4和9。相比而言,该工艺路线具有合成步骤短,副反应少,后处理简便,原料廉价易得,重复性高,安全和环境友好性等优点,具有较好的工业化前景。
[1] Roth G J, Binder R, Colbatzky F,etal. Nintedanib:From discovery to the clinic[J].J Med Chem,2015,58:1053-1063.
[2] Richeldi L, Costabel U, Selman M,etal. Eficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis [J].N Engl J Med,2011,365(12):1079-1087.
[3] Rieheldi L, du Bois R M, Raghu G,etal. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis [J].N Engl J Med,2014,370(22):2071-2082.
[4] Fukihara J, Kondoh T. Nintedanib(OFEV) in the treatment of idiopathic pulmonary fibrosis[J].Expert Rev Respir Med,2016,10(12):1247-1254.
[5] Keating G M.Nintedanib:A review of its use in patients with idiopathic pulmonary fibrosis[J].Drugs,2015,75(10):1131-1140.
[6] Rao T, Zhang C. Oxindole inhibitors of tyrosine kinase:WO 2015153877[P].2014.
[7] Merten J, Linz G, Schnaubelt J,etal. Process for the manufacture of an indolinone derivative:WO 2009071523[P].2009.
[8] Merten J, Renner S, Reichel C. Indolinone derivatives and process for their manufacture:WO 2009071524[P].2009.
[9] Roth G J, Heckel A, Colbatzky F,etal. Design,synthesis,and evaluation of indoli nones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone(BIBF 1120)[J].J Med Chem,2009,52:4466-4480.
[10] Roth G, Sieger P, Linz G,etal. 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition:WO 2004013099[P].2004.
[11] Tala S D, Ou T H, Lin Y W,etal. Design and synthesis of potent antitumor water-soluble phenylN-mustard-benzenealkylamide conjugatesviaa bioisostere approach[J].J Med Chem,2014,76:155-169.
[12] 谭君,陈碧微,高恩松,等. 一种6-甲氧羰基吲哚的制备方法:CN 201010189722[P].2010.
A New Synthetic Method of Nintedani
ZHAO Fei-yi, SHAO Ling-yan, GUO Ying*, JI Ya-fei*
(School of Pharmacy, East China of Science and Technology, Shanghai 200237, China)
N-methyl-4-nitro aniline was subjected to a sequential chloroacetylation, nucleophilic substitution and catalytic hydrogenation sequence achieving a key intermediateN-(4-aminophenyl)-N-methyl-2- (4-metrylpiperazin-1-yl) chloroacetamide(4). 6-Methoxycarbonyl-2-oxindole(8) was prepared from 4-chlorine-3-nitro benzoic acidviaa four-step reaction of esterification, nucleophilic substitution, catalytic hydrogenation and cyclization. Then, another key intermediate, methyl-1-(acetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate(9) was prepared by a “one-pot” process of compound8in the presence of triethyl orthobenzoate and acetic anhydride. Nintedanib with an overall yield of 57.2% was synthesizedviaa “one-pot” process of substitution of4with9and then deprotection. The structure was confirmed by1H NMR,13C NMR and MS(ESI).
N-methyl-4-nitro aniline; 4-chlorine-3-nitro benzoic acid; Nintedanib; idiopathic pulmonary fibrosis; tyrosine kinase inhibitor; one-pot; drug synthesis
2017- 02-09;
: 2017-07-17
国家自然科学基金资助项目(21476074, 21676088)
赵菲熠(1991-),女,汉族,浙江舟山人,硕士研究生,主要从事药物合成的研究。 E-mail: feiyizhao1230@163.com
郭瑛,博士, E-mail: zhiyaogy@163.com; 冀亚飞,教授, Tel. 021-64253314, E-mail: jyf@ecust.edu.cn
R914.5; O626.21
: ADOI: 10.15952/j.cnki.cjsc.1005-1511.2017.09.17022
