克唑替尼关键中间体(S)-1-(2,6-二氯-3-氟苯基)乙醇的不对称合成
2017-09-16李国贤罗宏军
张 杰, 李国贤, 乔 萍, 罗宏军, 梁 文, 薛 涛
(扬子江药业集团有限公司,江苏 泰州 225300)
·制药技术·
克唑替尼关键中间体(S)-1-(2,6-二氯-3-氟苯基)乙醇的不对称合成
张 杰*, 李国贤, 乔 萍, 罗宏军, 梁 文, 薛 涛
(扬子江药业集团有限公司,江苏 泰州 225300)
(S)-1-(2,6-二氯-3-氟苯基)乙醇(2)是合成抗癌药物克唑替尼的关键手性前体。本文以1-(2,6-二氯-3-氟苯基)乙酮为起始原料,利用二异松莰基氯化硼[(-)-Ipc2BCl]不对称还原制得光学纯的2;并将中间体2经Mitsunobu反应、还原、溴代、 Suzuki偶联及脱除Boc保护合成克唑替尼,其结构1H NMR,13C NMR和HR-MS(ESI)确证。对关键中间体2的合成条件进行了优化,并其对反应机理进行了推测。
2,6-二氯-3-氟苯乙酮; (S)-1-(2,6-二氯-3-氟苯基)乙醇; 克唑替尼; 二异松莰基氯化硼; 不对称还原; 药物合成
克唑替尼(Crizotinb)(1, Chart 1),化学名为3-[(R)-1-(2,6-二氯-3-氟苯基)乙氧基]-5-[1-(哌啶-4-基)-1H-吡唑-4-基]吡啶-2-胺,由美国辉瑞公司研制,于2011年获得美国FDA批准使用,是FDA第一个批准进行Ⅲ期临床实验的ALK酪氨酸激酶受体抑制剂。之后在多个国家(含中国)批准上市[1]。该药的发明及应用代表了个体化治疗的重大突破,是抗癌新药开发的又一个里程碑。2013年美国国立综合癌症网络(NCCN)与欧洲肿瘤内科学会(ESMO)都推荐克唑替尼作为ALK阳性非小细胞肺癌一线治疗的首选药物[2-4]。
克唑替尼的结构中含有苯环、吡啶环和吡唑环等3个芳香环。苯环与吡啶环通过醚键连接,吡唑环与吡啶环直接相连。克唑替尼的现有合成方法均是先用苯环片段与吡啶片段反应得到醚片段,再与吡唑片段经Suzuki偶联反应得到目标产物。合成的难点是如何得到关键手性中间体(S)-1-(2,6-二氯-3-氟苯基)乙醇(2, Chart 1)[5]。以2,6-二氯-3-氟苯乙酮为原料,目前关键中间体2的合成方法主要分为酶催化法[5-7]、不对称合成法[8-10]和手性拆分法[11]三种。但以上方法均存在一些缺陷,如不对称催化合成需要制备手性催化剂,其中过渡金属和手性配体价格昂贵,且反应条件相对苛刻;酶催化法反应条件要求较高,且酶的稳定性较差;手性拆分法步骤繁琐,收率较低等。

Scheme1
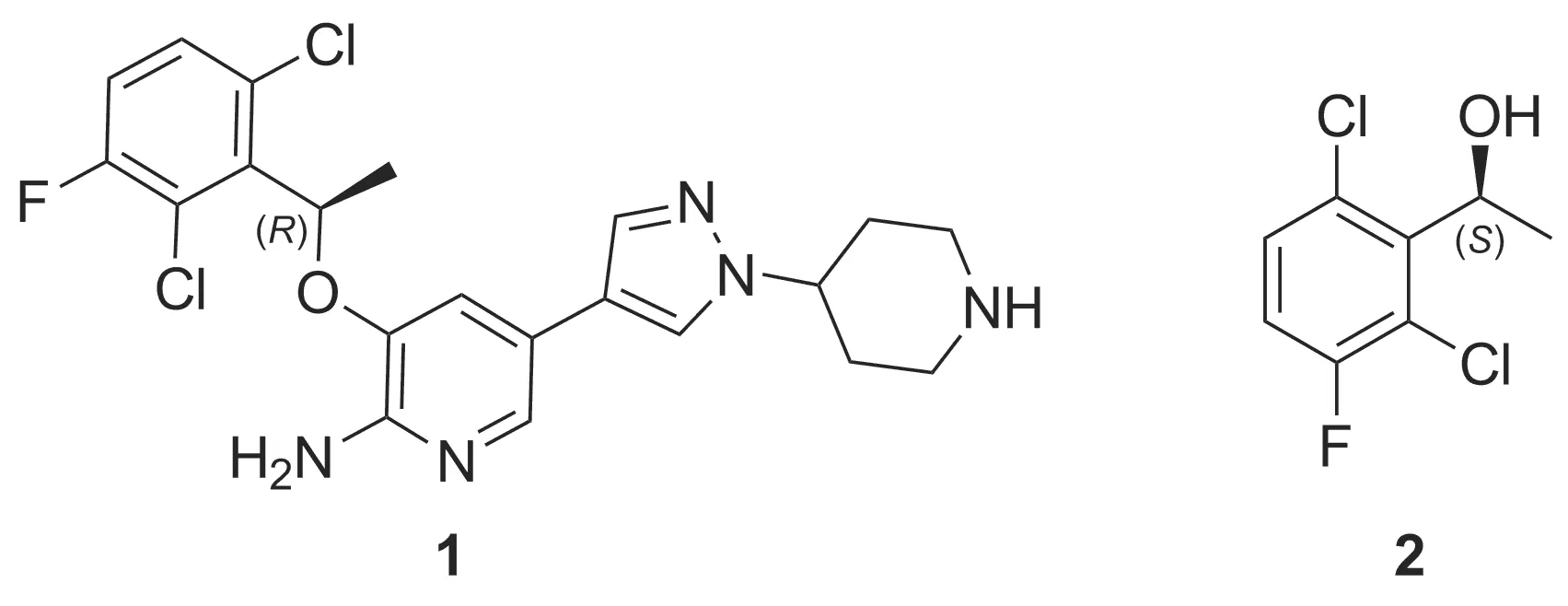
Chart 1
二异松莰基氯化硼[(-)-Ipc2BCl,3]属于α-蒎烯类手性硼烷还原剂,能有效还原芳基烷基酮,且对映选择性较高[12]。本文以2,6-二氯-3-氟苯乙酮(4)为原料,3为手性还原试剂,通过对反应条件优化,实现了对克唑替尼关键手性中间体2的不对称合成;中间体2经Mitsunobu反应、还原、溴代、Suzuki偶联并脱除Boc保护成功合成1(Scheme 1),其结构经1H NMR,13C NMR和HR-MS(ESI)确证。并对2的合成工艺进行了优化,在最佳工艺条件下,2收率及光学纯度较高,反应条件温和,适合工业化生产。
1 实验部分
1.1 仪器与试剂
Mettler MP90型熔点仪;MCP型旋光仪;Bruker 400核磁共振波谱仪(CDCl3为溶剂,TMS为内标);HR-ESI-MS Bruker APEXIII 7.0 TESLA FTMS型质谱仪;Agilent 1260型高效液相色谱仪(Water C18柱,5 μm, 150 mm×4.6 mm);Waters型高效液相色谱仪(手性Chiralpak AD-H柱);HPLC条件: 流动相为甲醇/水(V/V=70/30),流速为1 mL·min-1。
所用试剂均为化学纯。
1.2 合成
(1)2的合成
“小金鲤”篆刻社团诞生了一批优秀小“刀客”:在2017年的杭州市中小学生宪法主题书法篆刻作品评展中,袁润同学获得银奖,袁天奕、葛沁瑶同学获得铜奖,支子轩同学获得优秀奖;在2018年区艺术节篆刻比赛中支子轩、袁润、袁天奕、葛沁瑶、章志愿同学荣获五个一等奖,袁润同学的作品还参加了省艺术节展评。
(2)6的合成

(3)7的合成
将6120 g(0.36 mol)、铁粉100.8 g(0.89 mol)加至乙醇3 L中,搅拌下回流反应20 min;加入1 mol·L-1盐酸(109 mL),保温反应3 h。冷却至室温,用硅藻土助滤,滤饼用乙醇(2×300 mL)淋洗,合并滤液和洗液,减压蒸干得棕色固体(R)-3-(1-(2,6-二氯-3-氟苯基)乙氧基)吡啶-2-胺(7)109.1 g,收率98.3%; HR-MS(ESI)m/z: Calcd for C13H11N2OFCl2{[M+H]+}300.023 2, found 300.023 1。
(4)8的合成

(5)10的合成
氮气氛围下,将8110 g(0.29 mol)、 1-(N-Boc-4-哌啶基)-4-溴吡唑(9)131 g(0.35 mol)、四丁基溴化铵0.8 g(2.32 mmol)和碳酸铯282.8 g(0.87 mol)加至甲苯(700 mL)和水(600 mL)混合液中,搅拌下加入Pd(dppf)2CH2Cl21.8 g(2.32 mmol),于80 ℃反应4 h。冷却至室温,分液,有机相减压蒸除甲苯,残余物用800 mL甲苯/正庚烷(V/V=1/1)重结晶得类白色粉末3-[(R)-1-(2,6-二氯-3-氟苯基)乙氧基]-5-[1-(N-Boc-4-哌啶基)-1H-吡唑-4-基]-2-吡啶胺(10)124.4 g,收率78%; HR-MS(ESI)m/z: Calcd for C26H30N5O3FCl2{[M+H]+} 549.171 0, found 549.171 2。

2 结果与讨论
2.1 合成
由于1的分子结构中只含有一个手性中心,且是由关键手性中间体2引入,所以中间体2的光学纯度是整个工艺成功与否的关键因素。本文分别考察了反应溶剂及用量、反应温度、反应时间、碱的用量及还原剂用量对反应的影响,对手性中间体2的合成工艺进行了优化。
表1为溶剂及其用量对反应的影响。由表1可以看出,由于乙醇对原料4的溶解度不高,反应效果不理想,原料4剩余较多。二氯甲烷与四氢呋喃为溶剂时,反应均能顺利进行,且产物的ee值也较高,但是考虑到四氢呋喃的价格及套用处理,综合评估,选择二氯甲烷作为溶剂,保证工艺可行的同时,节约了成本。在考察溶剂用量时,发现降低溶剂二氯甲烷的用量,会影响原料4的转化及产物的ee值,故选择了二氯甲烷的用量为10 mL·g-1。

表1 溶剂及其用量对反应的影响*
*按原料4投料量换算溶剂用量。
表2为反应温度对反应的影响。可以看出,在一定温度范围内,温度越低,越有利于反应的进行,且产物的ee值也越高。由于(-)-Ipc2BCl对热不稳定,会分解为α-蒎烯,使反应不能顺利进行,所以在0~10 ℃,4的剩余量明显增多。在-25~0 ℃下反应,无论是4的剩余量及产物的ee值均在可接受范围之内,结合工业生产的实际操作及设备能耗成本等问题,确定反应温度为-5~0 ℃。

表2 反应温度对反应的影响Table 2 The effect of temperature on the reaction
表3为反应时间对反应的影响。由表3可见,在2~6 h内,随着反应时间的延长,有利于原料4的转化,且产物2的ee值也逐步提高,由于反应本身存在一定的平衡,所以反应原料4剩余到一定量后就很难再反应下去,即使延长反应时间也很难反应完,且延长反应时间会明显影响到产物的ee值,综合考虑,确定反应时间为6~8 h。
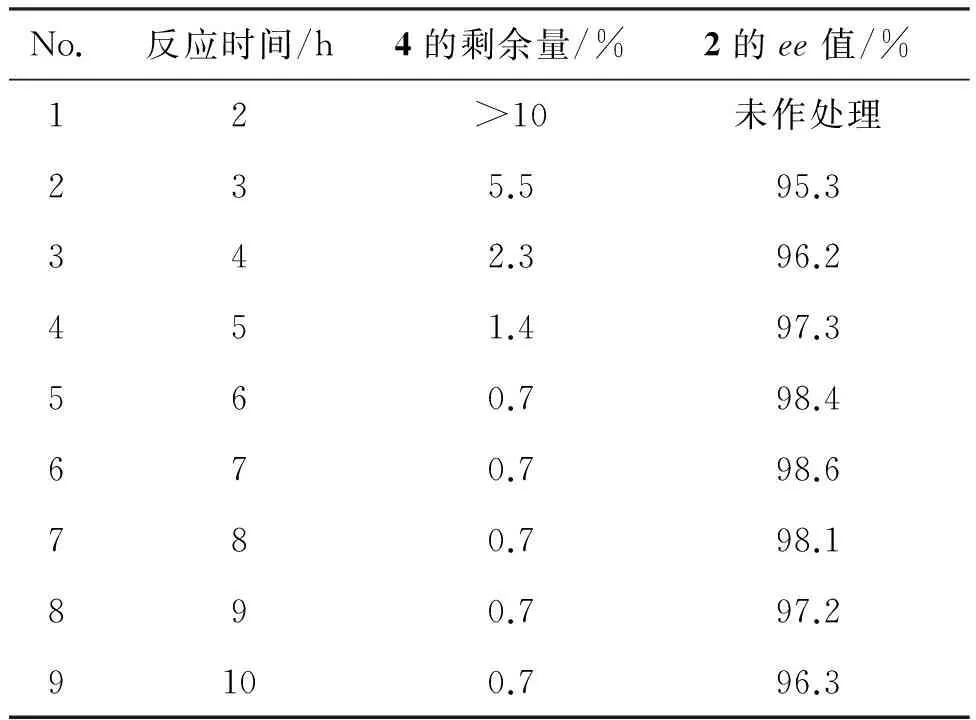
表3 反应时间对反应的影响
碱及其用量对反应的影响结果见表4。由表4可以看出,碱的加入明显促进了反应的进行,在一定的范围内,增加碱的用量可以提高4的转化及产物2的ee值。当碱的用量继续增大时,对反应有一定的抑制作用,原因可能是过量的碱影响了(-)-Ipc2BCl的稳定性。三乙胺与DIPEA效果相当,从成本角度出发,选择了三乙胺为碱,用量为0.5 eq.。
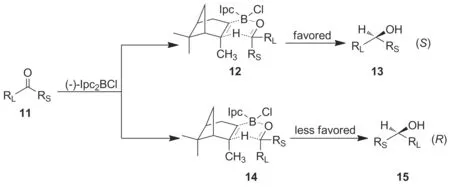
Scheme 2
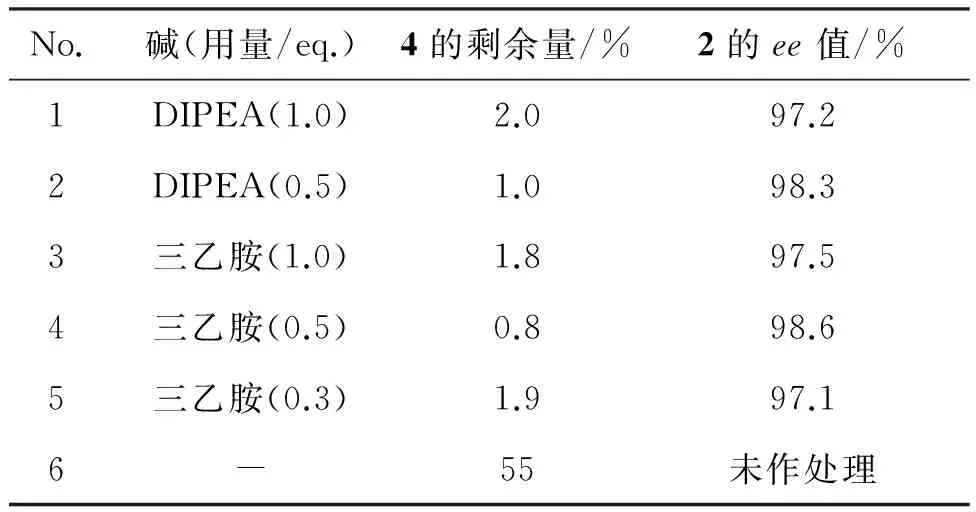
No.碱(用量/eq.)4的剩余量/%2的ee值/%1DIPEA(1.0)2.097.22DIPEA(0.5)1.098.33三乙胺(1.0)1.897.54三乙胺(0.5)0.898.65三乙胺(0.3)1.997.16-55未作处理
(-)-Ipc2BCl用量对反应的影响结果见表5。由表5可以看出,增加(-)-Ipc2BCl的用量,促进了反应的进行,(-)-Ipc2BCl用量为1.5 eq.时较为合适,无论是4的剩余量及产物2的ee值,均较为理想。在1.5~2.0 eq.范围内,加大(-)-Ipc2BCl的用量,会使整个反应体系变得复杂,4的剩余量并没有明显减少,且产品ee值有下降趋势。最终确定(-)-Ipc2BCl用量为1.5 eq.。
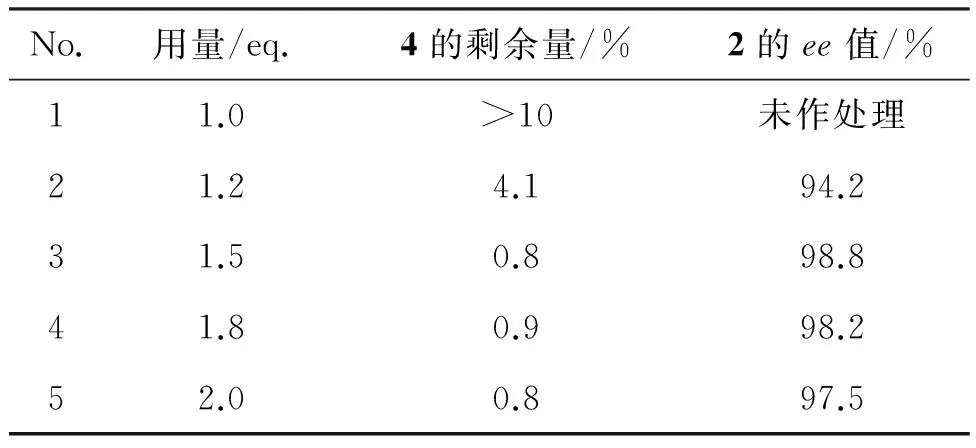
表5 (-)-Ipc2BCl用量对反应的影响Table 5 The effect of (-)-Ipc2BCl dosage on the reaction
通过对反应条件的优化,确定了最优工艺条件为:以二氯甲烷作为反应溶剂,三乙胺作碱性,其用量为0.5 eq.,还原剂(-)-Ipc2BCl的用量为1.5 eq.,反应温度为-5~0 ℃,反应时间在6~8 h左右,HPLC对反应过程进行中控,当原料4小于1.0%,即可停止反应后处理。以该工艺制得的中间体2为原料,可以成功合成1。
2.2 反应机理
对2的合成反应机理进行了推测:前手性酮11的氧与(-)-Ipc2BCl的硼配位,转化为刚性双六元环状过渡态12和14的平衡体系,受到基团空间位阻的影响,最终主要以优势构象12存在进而实现反应转化:发生分子内β-位的氢转移,并释放出α-蒎烯,实现羰基不对称还原为S构型产物13(Scheme 2)。
以(-)-Ipc2BCl为手性还原剂,通过对反应条件的优化,实现了对克唑替尼关键中间体(S)-1-(2,6-二氯-3-氟苯基)乙醇的不对称合成,获得了较为适应工业化生产的工艺路线。针对优化后的工艺,进行了中试放大,反应结果达到了预期目标,验证了工艺的可行性。
[1] 郭宗儒. 基于酶结构设计的个体化治疗药物克里唑替尼[J].药学学报,2016,51(4):672-676.
[2] Sano Y, Hashimoto E, Nakatani N,etal. Combining onartuzumab with erlotinib inhibits growth of non-small cell lung cancer with activating EGFR mutations and HGF overexpression[J].Mol Cancer Ther,2014,14(2):533-541.
[3] Toschi L, Cappuzzo F. Clinical implications of MET gene copy number in lung cancer[J].Future Oncol,2010,6(2):239-247.
[4] Pan Y, Zhang Y, Li Y,etal. ALK,ROS1 and RET fusions in 1139 lung adenocarcinomas:A comprehensive study of common and fusion pattern-specific clinic opathologic,histologic and cytologic features[J].Lung Cancer,2014,84(2):121-126.
[5] Martinez C A, Keller E, Meijer R,etal. Biotransformation-mediated synthesis of (1S)-1-(2,6-dichloro-3- fluorophenyl)ethanol in enantiomerically pure form[J].Tetrahedron:Asymmetry,2010,21(19):2408-2412.
[6] Cui J J, Tran D M, Shen H. Structure based drug design of crizotinib(PF-02341066),a potent and selective dual inhibitor of mesenchymal-epithelial transition factor(c-MET) kinase and anaplastic lymphoma kinase (ALK)[J].Journal of Medicinal Chemistry,2011,54(18):6342-6363.
[7] De K P D, Mcandrew D, Moore R. Fit -for-purpose development of the enabling route to crizotinib (PF-02341066)[J].Organic Process Research & Development,2011,15(5):1018-1026.
[8] 唐虹. 一种克唑替尼的制备方法:CN 102584795[P].2012.
[9] 沈立新,吴鹏程,刘福双,等. 一种制备c-Met抑制剂PF22341066的新方法:CN 101735198[P].2009
[10] Qian J Q, Yan P C, Che D Q. A novel approach for the synthesis of Crizotinib through the key chiral alcohol intermediate by asymmetric hydrogenation using highly active Ir-spiro-PAP catalyst[J].Tetrahedron Letters,2014,55(9):1528-1531.
[11] 路国梁,孙学英,齐放,等. 抗肿瘤靶向分子药物克里唑替尼的合成方法:CN 102532106[P].2012
[12] Remachandran P V, Pitre S, Brown H C. Selective reductions effective intramolecular asymmetric reductions ofα-,β-,andγ-Keto acids with diisopinocampheylborane and intermolecular asymmetric reductions of the corresponding esters with B-chlorodiisopinocampheylborane[J].Journal of Organic Chemistry,2002,67(15):5315-5319.
Synthesis of Crizotinib Key Intermediate (S)-1-(2,6-Dichloro-3-fluorophenyl)ethanol
ZHANG Jie*, LI Guo-xian, QIAO Ping, LUO Hong-jun, LIANG Wen, XUE Tao
(Yangtze River Pharmaceutical Group Co., Ltd., Taizhou 225300, China)
Using (-)-Ipc2BCl as the chair reduction agent, corresponding chiral precursor of Crizotinib. (S)-1-(2,6-dichloro-3-fluorophenyl) ethanol (2) was yielded by asymmetric reduction from 2,6-dichloro-3-fluro acetophenone. Based on key intermediate2, Crizotinib was synthesized. The process included Mitsunobu reaction, reduction of an arylnitro group, bromination reaction, Suzuki coupling and Boc deprotection. The structure of target compound was confirmed by1H NMR,13C NMR and HR-MS(ESI). The synthetic process of key intermediate2was optimized and the possible reaction mechanism was proposed.
2,6-dichloro-3-fluro acetophenone; (S)-1-(2,6-dichloro-3-fluorophenyl) ethanol; Crizotinib; (-)-Ipc2BCl; asymmetric reduction; drug synthesis
2016-11-30;
: 2017-07-18
张杰(1986-),男,汉族,江苏泰州人,硕士,主要从事药物合成的研究。 E-mail: zhang.jie@yangzijiang.com
R914.5; O621.3
: ADOI: 10.15952/j.cnki.cjsc.1005-1511.2017.09.16299
