两种香豆素分子与TiO2之间电荷转移的理论研究
2017-09-12吴文鹏李宁宁
吴文鹏,潘 霄,李宁宁,柴 云
(1.河南大学 化学化工学院,河南 开封 475004; 2.河南大学 环境与分析科学研究所,河南 开封 475004)
两种香豆素分子与TiO2之间电荷转移的理论研究
吴文鹏1,2,潘 霄1,2,李宁宁1,2,柴 云1*
(1.河南大学 化学化工学院,河南 开封 475004; 2.河南大学 环境与分析科学研究所,河南 开封 475004)
染料敏化太阳能电池以其低成本高效率引起了人们广泛的关注,其中的光诱导电荷转移过程对太阳能电池的光电转化效率起着重要作用. 本工作中我们以两种香豆素类染料分子7-羟基香豆素-4-乙酸(HCA)和7-N,N-二甲胺基香豆素-4-乙酸(DMACA)为例,从理论上研究了它们的几何结构和电子吸收光谱;并将其吸附在TiO2表面上,计算了它们与TiO2表面之间电荷转移的重组能、耦合强度和驱动力,进而计算了电荷转移速率. 结果表明,DMACA分子中二甲胺基在第一激发态的旋转能垒约为0.08 eV,因此DMACA分子在第一激发态时很容易发生扭转. 通过对HCA-TiO2/DMACA-TiO2体系中电子转移过程的研究,发现尽管两者重组能相似,但前者耦合强度和驱动力比后者小,使前者的电子转移速率略小于后者. 当DMACA-TiO2体系中二甲胺基在激发态发生扭转后,耦合强度略微减小,但由于驱动力减小,重组能增大,电子注入速率明显降低. 因此,本工作不仅合理地解释了实验现象,而且也提供了一种理论预测染料分子-半导体界面上电子转移的可行性方法.
香豆素;光诱导电荷转移;密度泛函理论;含时密度泛函理论
在过去的二十多年中,染料敏化太阳能电池以其低成本高效率成为人们研究的热点之一[1-6]. 这种太阳能电池通常将过渡金属配合物或者纯有机染料分子吸附在半导体表面上,如二氧化钛(TiO2). 在染料敏化太阳能电池中,光诱导电荷转移过程是一个主要的步骤,因为该步骤产生了光生自由电荷载流子. 染料分子吸附在TiO2团簇上是研究染料敏化太阳能电池的一个很好的模型[7-9],这方面的理论研究文献已有报道. 如SNCHEZ-DE-ARMAS等[10-14]用含时密度泛函理论(TD-DFT)研究了一系列的染料分子吸附在TiO2团簇上的电子结构和光学性质,这些染料分子包含儿茶酚、茜素、香豆素衍生物等. AGRAWAL等[15-16]同样用TD-DFT研究了香豆素和聚烯染料分子吸附在TiO2纳米颗粒表面上的光学性质. 然而,在这些理论计算中,大多数只考虑了前线轨道和垂直跃迁能,对其界面处电荷转移过程的研究相对较少.
香豆素类染料分子不含金属,是一类有应用前途的染料敏化剂. 7-羟基香豆素-4-乙酸(HCA)和7-N,N-二甲胺基香豆素-4-乙酸(DMACA)分子结构很相似,唯一的差别是香豆素环上的-OH被-N(CH3)2取代(图1),但实验上发现前者电子注入到TiO2纳米颗粒中的效率比后者高[17]. 作者认为是后者在激发态时产生了扭转的分子内电荷转移态(TICT),该态将电子注入TiO2导带的效率不高. 为了更深入地认识这一现象,本工作的目的就是用理论计算来研究HCA和DMACA分子与TiO2表面之间的电荷转移过程. 先前的研究[8,18-20]表明,DFT在优化分子的结构和预测分子光谱方面表现出色. 因此,我们采用DFT来研究HCA和DMACA的分子结构和光谱.
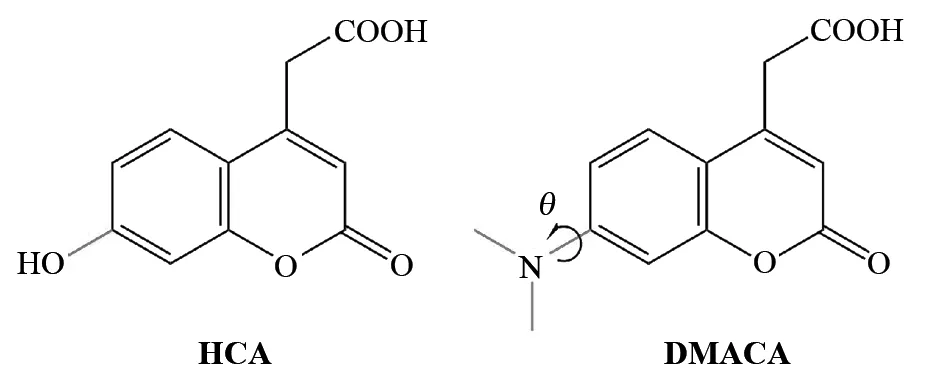
图1 HCA和DMACA的分子结构示意图Fig.1 Molecular structure of HCA and DMACA
1 计算方法
1.1 量子化学计算方法
HCA, DMACA, HCA-TiO2和DMACA-TiO2体系基态的几何结构用B3LYP泛函[21-22]优化,基组采用6-31+G**,金属Ti原子采用赝势基组LanL2dz,并进行红外振动频率分析. 垂直激发能用含时密度泛函TD-B3LYP[23-24],基组同前,水溶液的溶剂效应采用导体极化连续介质模型[24-25]. 所有量化计算都是在Gaussian09程序包[26]上完成.
1.2 电子转移速率的计算方法
染料分子到半导体表面的电子转移速率可由公式(1)计算[27-28]:
(1)
其中,Vdk是给体和表面各个电子态的电子耦合强度,可通过简化两态模型[8,29-31]进行估算;f() 是Fermi-Dirac分布,可表示为
(2)
其中,kB是Boltzman常数,T是热力学温度,Franck-Condon因子FC()为
(3)
其中,ΔG是受体态位于导带边时的自由能差,也称驱动力,λ为重组能. 在电荷转移过程中,总的重组能可以分为内重组能λi和外重组能λo,后者又常称为溶剂重组能. 驱动力可以通过Rehm-Weller方程[32]进行计算:
(4)
(5)
E(D+/D)为电子给体的氧化电势,E(A/A-)为受体的还原电势;ΔE00为给体0-0带跃迁能;Ecb为D+和A-之间的库仑作用能,可由式(6)估算[8]:
(6)
qi为第i个原子的电荷,rij是原子i和j之间的距离,ε0是真空电容率,εr为介质的相对介电常数.
该部分的计算是在量化计算获得参数的基础上用自编程序完成的.
2 结果与讨论
2.1 几何结构
优化得到的HCA和DMACA的分子结构见图2. 为了研究染料分子与TiO2间的相互作用,我们采用Ti9O18簇模型来模拟TiO2纳米颗粒,因已经有多篇文献用该簇模型来预测染料-半导体体系的光谱性质[8-14]. 我们采用双齿螯合模型模拟羧基和TiO2之间的相互作用,优化得到的结构见图2. 图2中标出了键合部位C-O和Ti-O键长. 从图2中可以看出,HCA和DMACA吸附在TiO2表面上后,羧基中C=O键长增长,C-O键长缩短,两个C-O键长趋于平均化;Ti-O键长在0.21~0.23 nm之间,比TiO2晶体中的略长. 从键角上看,O-C-O键角都从123°减小到118°. 从计算得到的结合能上看,HCA和DMACA与TiO2间的结合能分别为-0.74和-0.67 eV,前者略大.

键长单位:nm.图2 优化得到的各体系的分子结构球棍模型图Fig.2 Optimized molecular structures with ball and stick model
与HCA分子不同的是,DMACA分子中连接氮原子的两个甲基可以旋转(扭转角θ见图1). 通过改变θ进行部分优化扫描势能面,得到了基态和激发态的势能曲线(见图3). 从图3可以看出,θ=0.9°时能量最低,为基态最稳定构型;基态时旋转能垒为0.35 eV(优化得到了过渡态). 通过扫描激发态势能面,得到了两个稳定构型,θ分别为-3.0°和89.5°,后者的能量比前者低了约0.14 eV. 我们将前者称为分子内电荷转移态(ICT),后者称为扭转的分子内电荷转移态(TICT),估测ICT态到TICT态的旋转能垒约为0.08 eV(未优化过渡态). 因此在激发态时该分子很容易从ICT态变成TICT态,这与实验结果一致. 计算得到的ICT态和TICT态在水溶液中相对于基态的能量分别为3.04和2.90 eV,与实验值2.90和2.75 eV符合得很好[17]. 我们也对两个甲基旋转过程中发光的振子强度(f)进行了统计(见图4),发现θ在0°~50°之间f缓慢减小,在50°~60°之间f急剧降低,在60°~90°之间f又缓慢减小,在90°时f减为0,TICT态为非发射态,实验也证实了这一点[17].

图3 DMACA分子基态S0(上图)和激发态S1(下图)沿着扭转角的势能曲线Fig.3 Potential energy curves along torsion angle of the S0 ground state (top) and S1 excited state (bottom) of DMACA

图4 DMACA第一激发态振子强度随着扭转角的变化Fig.4 Plot of oscillator strength versus θ for S1 state of DMACA
在电荷转移过程中,给体和受体的能级对电荷转移至关重要,图5中我们画出了HCA、DMACA和Ti9O18簇以及它们形成的复合物的轨道能级图. 从图5我们可以看出,Ti9O18能级很密,几乎连成带;而且染料分子HCA、DMACA的LUMO能级都比Ti9O18的LUMO能级高,前者的HOMO能级也比后者的HOMO能级高. 从电子转移角度考虑,这将有利于电子从染料分子的LUMO能级向TiO2导带转移. 比较染料分子HCA和DMACA的能级可以发现,当-OH被-N(CH3)2取代后,HOMO和LUMO能级都升高了. 由此推测,当染料分子吸附在TiO2表面后,界面处电子转移的驱动力前者小于后者.

图5 计算得到的各体系的分子轨道能级Fig.5 Calculated molecular orbital energy levels of different systems
2.2 染料分子HCA和DMACA的电子吸收光谱和荧光发射光谱
图6中画出了理论模拟的HCA和DMACA分子在水溶液中的电子吸收光谱. 模拟光谱时采用高斯线型,峰半高半宽设为1 900 cm-1,相应光谱数据列于表1中. 从图6和表1可以看出,HCA和DMACA分别在315和375 nm处有一吸收峰,与实验值325和380 nm[17]相符,该吸收峰主要来自S0→S1的激发,对应HOMO→LUMO的跃迁,相应分子轨道轮廓图见图7. 从图7可以看出,HCA的HOMO主要分布在-OH和香豆素环上,LUMO主要分布在香豆素环上,-OH和-CH2COOH上也有较小分布;DMACA的HOMO主要分布在-N(CH3)2和香豆素环上,LUMO主要分布在香豆素环上,N原子和-CH2COOH上也有少量分布. 因此,HCA和DMACA HOMO→LUMO的跃迁都带有分子内电荷转移(ICT)的特征,与香豆素C343、D1421[8]类似. 除了该吸收峰外,我们还发现HCA和DMACA在较短波长处也存在较强吸收,这些吸收峰都是由较多跃迁叠加而成,详见图6和表1,这里不再一一赘述. 由于实验上只测了波长大于300 nm的光谱,实验上未观测到这些吸收峰. 当DMACA的二甲氨基发生扭转后,HOMO主要分布在-N(CH3)2上,苯环上也有少量分布,LUMO主要分布在香豆素环和-CH2COOH上(见图7),HOMO→LUMO的跃迁有明显的ICT的特征.
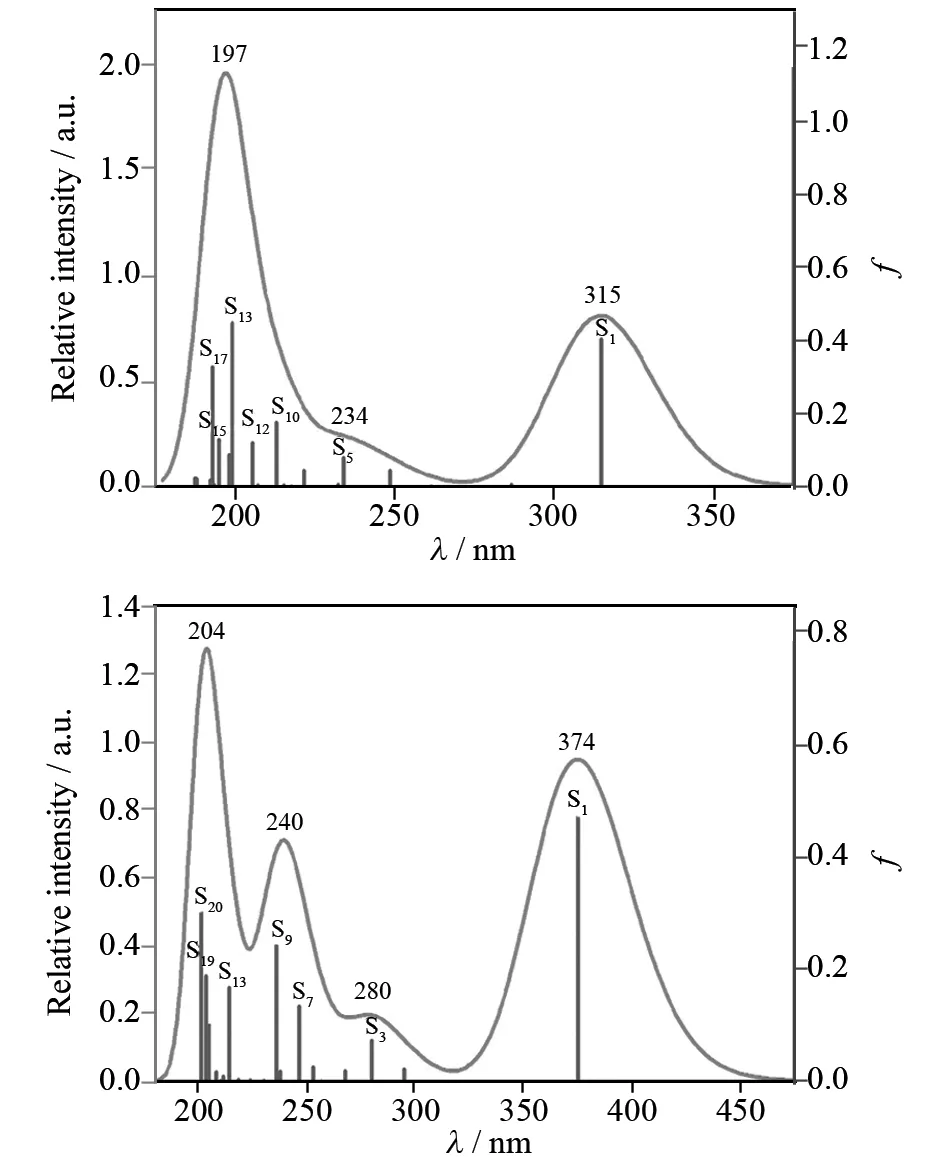
曲线为拟合所得光谱,竖线为计算得到的振子强度f.图6 理论拟合得到的HCA(上图)和DMACA(下图)的电子吸收光谱Fig.6 Theoretically fitted electronic absorption spectra of HCA(top) and DMACA(bottom)
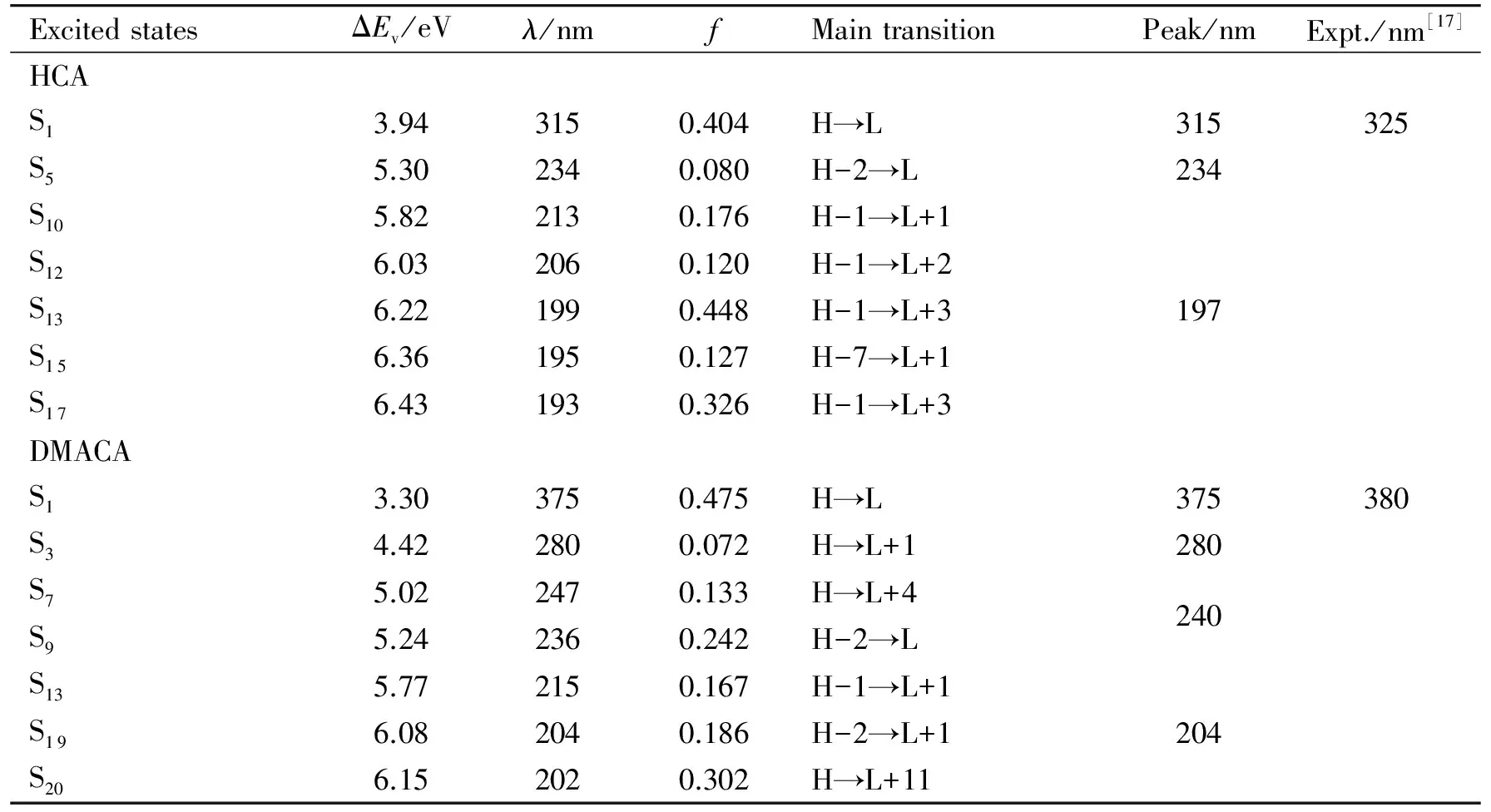
表1 计算得到的HCA和DMACA的垂直激发能a
aH代表HOMO,L代表LUMO,Peak指计算拟合得到的吸收峰的位置,Expt.为实验值.
同时我们对HCA和DMACA分子在水溶液中的垂直发射能进行了计算,它们的荧光发射峰分别出现在368和432 nm,对应的振子强度分别为0.584和0.596,表现出较强的发射特征. 对于DMACA-TICT态,发射峰位置在647 nm,振子强度为0,不发射荧光.

图7 ICT态和TICT态发生跃迁的轨道图Fig.7 Molecular orbitals of ICT state and TICT state
2.3 电子转移速率
在计算电子转移速率时,需要首先知道三个重要参数:重组能,耦合强度和驱动力. 为方便描述,染料分子简记为D,Ti9O18简记为A.
2.3.1 重组能
溶剂重组能可由公式(7)[33]估算,
(7)
Δe为给体转移给受体的电荷,rD和rA为给体D和受体A的半径,R为给体和受体中心间的距离,εop为介质的光学介电常数,等于折射率的平方;εs为静电介电常数.
对HCA-Ti9O18/DMACA-Ti9O18体系在水溶液中的溶剂重组能,我们采用的参数如下:rD=0.405/0.430 nm,rA=0.466 nm,R=0.871/0.896 nm,εop=1.778,εs=78.355. 利用公式(7),可预测溶剂的重组能为0.918/0.886 eV. 在计算D*A→D+A-电荷转移过程的内重组能时,我们将体系分成两部分,D*→D+和A→A-,然后分别进行计算. 表2列出了计算得到的D-A体系的重组能. 从表2可以看出,Ti9O18簇的重组能很大,为0.739 eV,表明这样一个簇的体系并不是一个很好的接受电子的体系(可能是因为簇选得太小,加入一个电子后,结构改变比较大). 总重组能的计算结果表明,HCA-Ti9O18和DMACA-Ti9O18体系的重组能几乎一样,DMACA-TICT-Ti9O18体系的重组能较大.
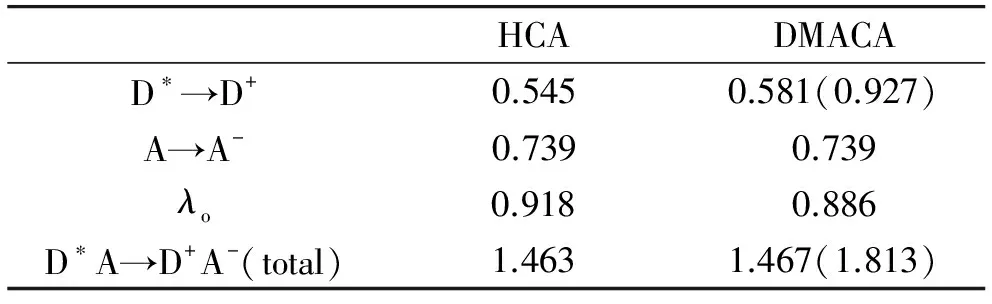
表2 各体系的重组能(单位:eV)
括号内数据为TICT态的计算值;总重组能列出的是忽略了Ti9O18簇的重组能的结果.
2.3.2 耦合强度
由于在Ti9O18簇的空能级挨得非常近(图5),我们计算了一系列Ti9O18簇的空轨道和染料分子LUMO之间的耦合强度,计算结果画于图8和图9中. 同时,我们也计算了DMACA体系TICT态的耦合强度,结果画于图10中. 从图8-图10可以看出, DMACA-ICT-Ti9O18体系的耦合强度最大,DMACA-TICT-Ti9O18体系的耦合强度比前者略小,HCA-Ti9O18体系的耦合强度最小. 因此,尽管HCA和DMACA与TiO2之间连接基团都是羧基,它们之间的耦合强度还是有差别的.

图8 计算得到的HCA-Ti9O18体系电子转移的耦合强度Fig.8 Calculated electronic transfer coupling strength of HCA-Ti9O18

图9 计算得到的DMACA-ICT-Ti9O18体系电子转移的耦合强度Fig.9 Calculated electronic transfer coupling strength of DMACA-ICT-Ti9O18

图10 计算得到的DMACA-TICT-Ti9O18体系电子转移的耦合强度Fig.10 Calculated electronic transfer coupling strength of DMACA-TICT-Ti9O18
2.3.3 驱动力
计算得到的HCA和DMACA分子激发态到半导体导带底的电子转移过程的驱动力列于表3中. 由于我们计算的是在水溶液中的驱动力,水的静电介电常数很大,由公式(6)可得库仑作用能很小,这里计算的驱动力忽略了该部分能量. 从表3可以看出,DMACA-ICT态到TiO2的驱动力最大,HCA的最小,这与它们的LUMO能级顺序一致.
2.3.4 电子转移速率
将以上三个参数带入方程(1),可得到电子转移的速率. 如果考虑A→A-过程的重组能,计算出的电子注入时间τ> 1 ps,与实验值100 fs[17]相差较大. 如果忽略该重组能,计算结果列于表3中. 从表3中可以看出电子转移的速率非常快,在HCA和DMACA中都达到了1013s-1数量级,相应的注入时间分别为79和45 fs,与实验文献报道的注入时间100 fs[17]接近;但电子从DMACA-TICT态注入TiO2速率较慢,这也与实验预测[17]一致. 究其原因,当二甲胺基扭转后,耦合强度和驱动力都略微减小,重组能增大较多,最终导致电子转移的速率降低. 尽管电子从DMACA-ICT态到TiO2的转移速率比HCA的大,但DMACA-TICT态能量较低,存在较大的分布,从整体上看,最终导致从DMACA电子转移的效率不及HCA的.

表3 各体系电子转移的驱动力和速率
3 结论
用密度泛函理论和含时密度泛函理论研究了香豆素HCA和DMACA分子的几何结构、电子吸收光谱和荧光发射光谱;在此基础上,计算了它们与TiO2表面之间的重组能、耦合强度和驱动力,进而计算了电荷转移速率. 得到的结论如下:
1) DMACA分子中二甲胺基在基态和第一激发态的旋转能垒分别为0.35 和0.08 eV,因此DMACA分子在第一激发态时很容易发生旋转,从ICT态转变为能量较低的TICT态,使激发态分子在TICT态有较多分布.
2) 计算得到HCA和DMACA在水溶液中的电子吸收光谱的第一个吸收峰分别在315和375 nm,与实验光谱325和380 nm符合较好.
3) 计算HCA-TiO2/DMACA-TiO2体系中电子转移速率的过程中忽略了库仑作用能和表面得到电子后的重组能,计算得到的电子转移速率都在1013s-1数量级,与文献报道的注入时间~100 fs一致. 尽管两者重组能相似,但前者耦合强度和驱动力比后者小,使前者的电子转移速率略小于后者. 当DMACA-TiO2体系中二甲胺基在激发态发生扭转后,耦合强度略微减小,但由于驱动力减小,重组能增大,电子注入速率明显降低.
因此,本工作不仅合理地解释了实验现象,而且也提供了一种理论预测界面上电子转移的可行性方法.
[2] HAGFELDT A, BOSCHLOO G, SUN L, et al. Dye-sensitized solar cells [J]. Chemical Reviews, 2010, 110: 6595-6663.
[3] CLIFFORD J N, MARTNEZ-FERRERO E, VITERISI A, et al. Sensitizer molecular structure-device efficiency relationship in dye sensitized solar cells [J]. Chemical Society Reviews, 2011, 40: 1635-1646.
[4] KAKIAGE K, AOYAMA Y, YANO T, et al. Highly-efficient dye-sensitized solar cells with collaborative sensitization by silyl-anchor and carboxy-anchor dyes [J]. Chemical Communications, 2015, 51: 15894-15897.
[5] YANG L, CHEN S, ZHANG J, et al. Judicious engineering of a metal-free perylene dye for high-efficiency dye sensitized solar cells: the control of excited state and charge carrier dynamics [J]. Journal of Materials Chemistry A, 2017, 5: 3514-3522.
[6] RICHHARIYA G, KUMAR A, TEKASAKUL P, et al. Natural dyes for dye sensitized solar cell: A review [J]. Renewable and Sustainable Energy Reviews, 2017, 69: 705-718.
[7] DUCAN W R, PREZHDO O V. Theoretical studies of photoinduced electron transfer in dye-sensitized TiO2[J]. Annual Review of Physical Chemistry, 2007, 58: 143-184.
[8] 吴文鹏. 有机太阳能电池中有机分子的光谱和电荷转移的理论研究[D]. 厦门: 厦门大学, 2013: 79-104.
WU W P. Theoretical investigations on the spectra and charge transfer of organic molecules in organic solar cells [D]. Xiamen: Xiamen University, 2013: 79-104.
[9] YANG L Z, WU W P, ZHAO Y. Effect of TiO2particles on normal and resonance Raman spectra of coumarin 343: a theoretical investigation [J]. Physical Chemistry Chemical Physics, 2015, 17: 10910-10918.
[15] AGRAWAL S, DEV P, ENGLISH N J, et al. First-principles study of the excited-state properties of coumarin-derived dyes in dye-sensitized solar cells [J]. Journal of Materials Chemistry, 2011, 21: 11101-11108.
[16] AGRAWAL S, DEV P, ENGLISH N J, et al. A TD-DFT study of the effects of structural variations on the photochemistry of polyene dyes [J]. Chemical Science, 2012, 3: 416-424.
[17] RAMAKRISHNA G, SINGH A, PALIT D K, et al. Effect of molecular structure on interfacial electron transfer dynamics of 7-N,N-dimethyl coumarin 4-acetic acid (DMACA) and 7-hydroxy coumarin 4-acetic acid (HCA) sensitized TiO2and ZrO2nanoparticles [J]. The Journal of Physical Chemistry B, 2004, 108: 12489-12496.
[18] 李云飞, 王新收, 吴文鹏. 一种基于苯并噻二唑衍生物的F-荧光探针分子的理论研究[J]. 化学研究, 2015, 26(6): 575-578.
LI Y F, WANG X S, WU W P. Theoretical investigations on a F-fluorescent probe based on benzotiadiazole derivative [J]. Chemical Research, 2015, 26(6): 575-578.
[19] 马原, 左桂云, 吴文鹏. 理论研究连接方式对低聚(3-己基噻吩)结构和电子光谱的影响[J]. 化学研究, 2016, 27(4): 450-454.
MA Y, ZUO G Y, WU W P. Effect of coupling ways on the structures and electronic spectra of oligo(3-hexylthiophene): A theoretical investigation [J]. Chemical Research, 2016, 27(4): 450-454.
[20] 安贝贝, 朱秋玲, 吴文鹏. 三种黄酮类化合物的电子吸收光谱的理论研究[J]. 化学研究, 2016, 27(6): 693-697.
AN B B, ZHU Q L, WU W P. Theoretical studies on the electronic absorption spectra of three flavonoids [J]. Chemical Research, 2016, 27(6): 693-697.
[21] BECKE A D. Density-functional thermochemistry. III. The role of exact exchange [J]. The Journal of Chemical Physics, 1993, 98(7): 5648-5652.
[22] LEE C, YANG W, PARR R G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density [J]. Physical Review B, 1988, 37: 785-789.
[23] FURCHE F, AHLRICHS R. Adiabatic time-dependent density functional methods for excited state properties [J]. The Journal of Chemical Physics, 2002, 117: 7433-7447.
[24] SCALMANI G, FRISCH M J, MENNUCCI B, et al. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model [J]. The Journal of Chemical Physics, 2006, 124: 094107.
[25] COSSI M, REGA N, SCALMANI G, et al. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model [J]. Journal of Computational Chemistry, 2003, 24: 669-681.
[26] FRISH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09 [CP]. Revision A.02, Wallingford CT: Gaussian, Inc., 2009.
[27] GAO Y Q, GEORGIEVSKII Y, MARCUS R A. On the theory of electron transfer reactions at semiconductor electrode/liquid interfaces [J]. The Journal of Chemical Physics, 2000, 112: 3358-3369.
[28] GOSAVI S, GAO Y Q, MARCUS R A. Temperature dependence of the electronic factor in the nonadiabatic electron transfer at metal and semiconductor electrodes [J]. Journal of Electroanalytical Chemistry, 2001, 500: 71-77.
[29] SENTHILKUMAR K, GROZEMA F C, GUERRA C F, et al. Absolute rates of hole transfer in DNA [J]. Journal of the American Chemical Society, 2005, 127: 14894-14903.
[30] VALEEV E F, COROPCEANU V, DA SILVA FILHO D A, et al. Effect of electronic polarization on charge-transport parameters in molecular organic semiconductors [J]. Journal of the American Chemical Society, 2006, 128: 9882-9886.
[31] ZHANG W W, LIANG W Z, ZHAO Y. Non-Condon effect on charge transport in dithiophene-tetrathiafulvalene crystal [J]. The Journal of Chemical Physics, 2010, 133: 024501.
[32] REHM D, WELLER A. Kinetics of fluorescence quenching by electron and H-atom transfer [J]. Israel Journal of Chemistry, 1970, 8: 259-271.
[33] MARCUS R A. On the theory of electron-transfer reactions. VI. Unified treatment for homogeneous and electrode reactions [J]. The Journal of Chemical Physics, 1965, 43: 679-701.
[责任编辑:刘红玲]
Theoretical studies on the charge transfer process between two coumarins and TiO2
WU Wenpeng1,2, PAN Xiao1,2, LI Ningning1,2, CHAI Yun1*
(1.CollegeofChemistryandChemicalEngineering,HenanUniversity,Kaifeng475004,Henan,China;2.InstituteofEnvironmentalandAnalyticalSciences,HenanUniversity,Kaifeng475004,Henan,China)
Dye-sensitized solar cells (DSSCs) have attracted extensive attention due to low cost and relative high efficiency. The photo induced charge transfer process is very important to the efficiency of DSSCs. In this work, we take two coumarins, i.e., 7-hydroxy coumarin 4-acetic acid (HCA) and 7-N,N-dimethyl amino coumarin 4-acetic acid (DMACA) as examples, and investigate their geometries and electronic absorption spectra theoretically. Furthermore, the three parameters controlling electron transfer rate, i.e., the reorganization energy, electronic coupling strength, and driving force, between the two coumarins and TiO2, are calculated to obtain electron transfer rate. It shows that rotation barrier of -N(CH3)2group in DMACA in the first excited state is predicted to be about 0.08 eV. So this group is very easy to twist. For HCA-TiO2and DMACA-TiO2systems, the reorganization energy is similar, but the electronic coupling strength and driving force in the former are smaller than those in the latter. As a result, the electron transfer rate in the former is a little smaller than that in the latter. When -N(CH3)2group in DMACA-TiO2system rotates, the electronic coupling strength decreases slightly, and the driving force decreases, and the reorganization energy increases, leading to a drastically decrease in the electron injection rate. Therefore, this work not only explains the experimental phenome-na, but also provides one practicable approach to predict the electron transfer process between dye molecules and semiconductors.
coumarin; photo induced charge transfer; density functional theory; time-dependent density functional theory
2017-06-18.
河南大学科学研究基金(2015YBZR009),河南大学博士科研启动基金(B2013141).
吴文鹏(1984-), 男, 讲师, 研究方向为理论与计算化学.*
, E-mail:chaiyun@henu.edu.cn.
O641
A
1008-1011(2017)04-0424-08
