青年型帕金森病1例报告并文献复习
2017-09-11杨宇宏袁宝玉张志珺
杨宇宏袁宝玉张志珺
·病例报告·
青年型帕金森病1例报告并文献复习
杨宇宏*袁宝玉*张志珺*
青年型帕金森病 Parki n基因 PLA2G6基因
帕金森病(Parkinson Disease,PD)是常见的中枢神经系统变性疾病,发病年龄多在50岁以上,50岁以前发病称为早发性帕金森病 (early-onset Parkinsonism,EOP),分为青少年型帕金森病 (21岁前起病)和青年型帕金森病(21岁至 50岁间起病,young-onset Parkinson disease, YOPD)[1]。现报告1例Parkin和PLA2G6基因联合突变的YOPD患者,并复习相关文献,以期提高对该病的认识。
1 临床资料
1.1 发病情况患者,女,32岁,心理科医生。因“渐起情绪差、眠差、运动迟缓2年余”于2017年1月3日入院。患者于2014年5月因家庭矛盾出现情绪差,兴趣缺乏,入睡困难,有自弃想法,自服“百忧解、思诺思”等药物,情绪及夜眠稍改善。后患者渐出现运动迟缓,右下肢行走时拖曳,骑车时自觉调整方向困难致向右偏斜。2014年12月患者行走不稳加重,起床、转身等动作启动迟缓,四肢僵硬,双上肢静止时不自主抖动,右上肢为重,表情减少。伴便秘,多汗,口干,无嗅觉减退。既往史(-)。个人史:2015年1月新房装修后直接入住,大学期间大量饮用咖啡(偶达7包/d)。家族遗传病史(-),父母非近亲结婚。
1.2 体格检查内科系统(-)。关期神经系统:神志清,精神可,高级认知正常。面具脸,慌张步态。颅神经(-)。四肢肌力5级,双上肢见静止性震颤,双上肢呈齿轮样肌张力增高,右上肢为著,双下肢呈铅管样肌张力增高,除右下肢膝反射(++++)外,余腱反射(+++),双侧踝阵挛(+),病理反射(-)。感觉系统(-)。共济试验(-)。脑膜刺激征(-)。开期神经系统:除四肢不自主舞动,四肢肌张力正常,双侧踝阵挛(-),无面具脸、慌张步态外,余查体与关期一致。
1.3 辅助检查①三大常规、血生化全套、肿瘤、炎症、风湿免疫、病毒八项、铜蓝蛋白(-),K-F环(-)。②头颅 MRI(-)。③简易精神状态检测量表30分,汉密尔顿抑郁量表14分,汉密尔顿焦虑量表19分,UPDRS运动评分 (关/开):72/2分;Hoehn-Yahr评分(关/开):5/2期。④PET检查(11C-CFT):双侧尾状核放射性分布轻度减少,双侧壳核前部和后部放射性摄取减少,以左侧明显。⑤PET检查(18F-FDG):双侧壳核放射性摄取增高,右侧略明显。⑥围绕导致运动障碍的基因行新一代测序+Sanger测序:患者Parkin基因外显子10重复变异,PLA2G6基因复合杂合突变 [c.1771C>T(p.Arg591Trp)杂合突变及 c.956C>T(p. Thr319Met)杂合突变],其中 c.1771C>T(p.Arg591Trp)来自母亲,c.956C>T(p.Thr319Met)来自父亲。
1.4 诊断、治疗及随访2015年3月起行左旋多巴试验性治疗,“美多芭62.5 mg 3次/d”后症状明显改善,诊断为PD,治疗方案为“普拉克索0.375 g 3次/d,美多芭62.5 mg 3次/d、司来吉兰5 mg 2次/d”。2月余后出现异动症及开关现象,调整药量疗效欠佳。2016年9月更换方案“息宁0.875 g/d,普拉克索0.25 g 4次/d,金刚烷胺100 mg 4次/d”后,起效减慢但异动症减少。由于自觉四肢僵硬感明显,2016年12月起患者自行在原有方案上加用“美多芭125 mg 2次/d”,起效时间未改善,且异动症及剂末现象加重。入院后行左旋多巴冲击试验发现,125 mg美多芭无效,250 mg美多芭的最大改善率为97.2%,但异动症严重。更换方案为“美多芭125 mg 5次/d,恩他卡朋0.2 g 5次/d,罗匹尼罗2 mg 1次/d”后,起效时间缩短,药效维持时间延长,但仍存在异动症。
2 讨论
YOPD病因普遍认为是基因、表观遗传和环境的相互作用[1]。目前已发现26种单基因突变形式,分为常染色体显性遗传 (如SNCA基因等)、常染色体隐性遗传 (如Parkin、PLA2G6基因等)和X染色体连锁遗传 (如ATP6AP2基因等)[2-4]。
1973年日本首次报告常染色体隐性遗传青少年型帕金森病 (autosomal recessive juvenile parkinsonism,AR-JP),该病特点为①起病年龄早,常于40岁前起病;②疾病进展缓慢;③下肢肌张力障碍,尤以足部肌张力障碍常见;③皮质脊髓束受损致下肢腱反射活跃;④嗅觉保留,痴呆少见(<3%),伴有精神症状;⑤左旋多巴反应性好,但左旋多巴诱导的症状波动及运动障碍出现早且频繁;⑥路易小体缺失[5]。1998年KITADA等[6]发现AR-JP由PARKIN基因突变所致,被命名为PARK2。Parkin基因位于6q25.2-q27,编码产生Parkin蛋白。Parkin基因突变为AR遗传,突变类型为点突变和外显子重排突变。纯合和复合杂合突变临床表型除可表现为AR-JP外,可表现为散发性EOP,其主要具备运动症状(即运动迟缓、肌强直、静止性震颤及姿势平衡异常),不典型症状如锥体束征、肌张力障碍等少见,且可出现嗅觉缺失,存在路易小体等[7];Parkin基因大片段重排杂合突变导致PD亦有报告,其临床特点多表现为上述散发性EOP[8-9]。
2009年PAISAN-RUIZ等[10]首次报告肌张力障碍-帕金森综合征 (dystonia-parkinsonism,DP)的家系中存在PLA2G6基因纯合点突变,被命名为PARK14。PLA2G6基因位于22q13.1,编码产生磷脂酶A2 VI型(iPLA2-VIA)。PLA2G6基因突变为AR遗传,具有高度基因多效性。与PD相关的突变类型是点突变,临床表型分为 DP、常染色体隐性遗传早发性帕金森综合征 (autosomal recessive early-onset parkinsonism,AREP)和EOP。DP多在20岁左右起病,首发症状可为自主神经功能障碍、精神心理障碍和认知障碍,具有典型运动症状和肌张力障碍,可伴有锥体束征、构音障碍、眼动异常和共济失调,早期对左旋多巴反应好,但疗效迅速减退并伴有严重异动症[11]。AREP多见于近亲结婚家系或存在家族史,中年起病,仅表现为单纯的PD[12]。EOP非近亲结婚家系和阴性家族史,可为杂合突变,青年起病,除典型运动症状外,还可存在认知障碍、共济失调和精神症状,但无肌张力障碍和锥体束征[13]。本例女性患者,青年起病,缓慢进展性病程,阴性家族史,父母非近亲结婚。首发症状为非运动症状,运动症状中首发非对称性运动迟缓,后出现其他运动症状、自主神经功能障碍和锥体束征,无嗅觉异常和认知下降;左旋多巴显效,极早出现运动并发症;基因检测显示Parkin基因外显子10重复变异(约1.7倍重复,为杂合重复突变)和PLA2G6基因复合杂合突变,故确诊为YOPD。
本例是首次报告的Parkin和PLA2G6基因联合突变的YOPD患者,同时具备上述两种基因突变的临床特点,但存在不一致之处,原因可能为两种基因突变作用叠加。Parkin基因外显子10重复变异主要影响Parkin蛋白环指样模体间区,可能进而影响其E3泛素蛋白连接酶活性[14]。PLA2G6基 因 c.1771C>T (p.Arg591Trp)/c.956C>T(p. Thr319Met)属于错义突变,查询HGMD和PDMutDB突变数据库后发现c.956C>T(p.Thr319Met)尚无报告,该突变位点位于锚蛋白重复区域,其作用为促进iPLA2-VIA发生低聚反应产生酶活性,因此该位点突变可能影响蛋白功能[9,13],Polyphen-2和MutationTaster软件均预测该变异很可能为致病突变。c.1771C>T(p.Arg591Trp)已被收录,WU等[11]报告c.1771C>T(p.Arg591Trp)/c.116G>T(p.R39Q)复合杂合突变表现为婴儿神经轴索变性,而本例导致PD,符合该基因的基因多效性特点。
本例患者另一特点为治疗后迅速出现运动并发症,原因可能为Parkin和PLA2G6基因突变作用叠加。这一现象提示对于早期出现运动并发症的YOPD患者,需警惕两种及以上基因突变。此外,治疗存在遗传因素的YOPD患者时应对左旋多巴延迟应用和控制剂量;对于已出现运动并发症的YOPD患者,应依据指南调整方案,必要时深部脑刺激治疗[15]。
综上所述,Parkin和PLA2G6基因突变的YOPD临床表现除运动症状和非运动症状外,还可存在锥体束征、共济失调和肌张力障碍等,临床上需提高认识。对于不典型YOPD患者及其家族成员,完善PD相关基因检测、甚至全基因组扫描十分必要。对于存在遗传因素的YOPD患者,治疗时应警惕运动并发症。
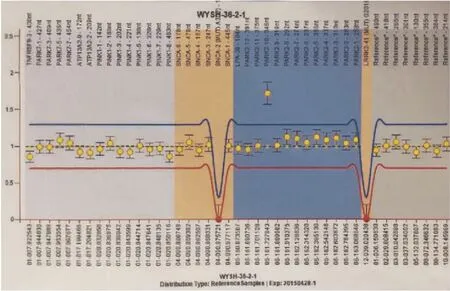
图1 Parki n基因测序图发现外显子10重复变异
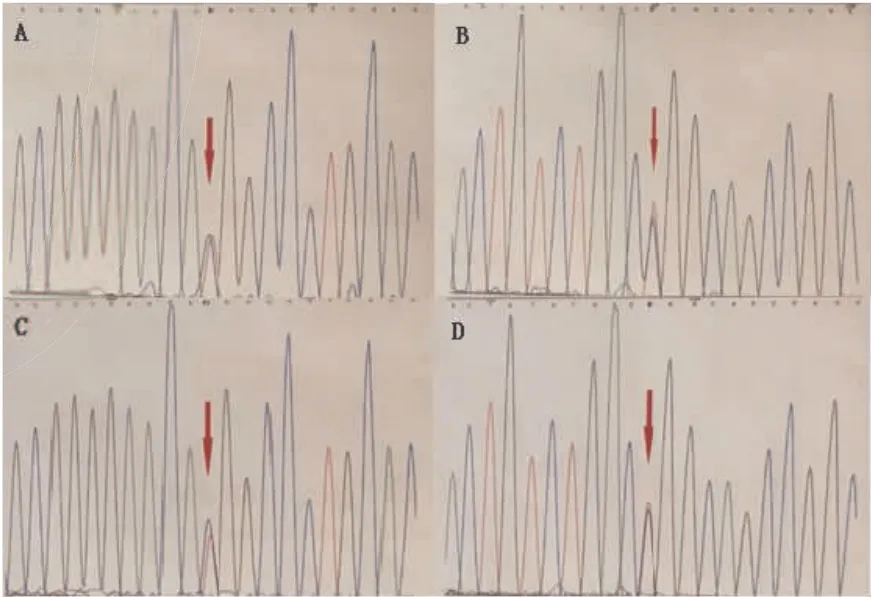
图2 PLA2G6基因测序图发现c.956C>T(p.Thr319M et)/c.1771C>T (p.Arg591Trp)复合杂合突变(箭头所示)。A:患者c.956C>T(p. Thr319M et)突变;B:患者c.1771C>T(p.Arg591Trp)突变;C:患者父亲c.956C>T(p.Thr319M et)突变;D:患者母亲c.1771C>T(p.Arg591Trp)突变
[1]SCHRAG A,SCHOTT JM.Epidemiological,clinical,and genetic characteristics of early-onset parkinsonism[J].Lancet Neurol,2006,5(4):355-363.
[2]FUNAYAMA M,OHE K,AMO T,et al.CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease:a genomewide linkage and sequencing study[J].Lancet Neurol,2015,14 (3):274-282.
[3]VERSTRAETEN A,THEUNS J,VAN BROECKHOVEN C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era[J].Trends Genet,2015,31(3):140-149.
[4]WILSON GR,SIM JC,MCLEAN C,et al.Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with alpha-synuclein pathology[J].Am J Hum Genet,2014,95(6):729-735.
[5]KHAN NL,BROOKS DJ,PAVESE N,et al.Progression of nigrostriatal dysfunction in a parkin kindred:an [18F]dopa PET and clinical study[J].Brain,2002,125(Pt 10):2248-2256.
[6]KITADA T,ASAKAWA S,HATTORI N,et al.Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism[J]. Nature,1998,392(6676):605-608.
[7]HOULDEN H,SINGLETON AB.The genetics and neuropathology of Parkinson's disease[J].Acta Neuropathol,2012,124(3): 325-338.
[8]MACEDO MG,VERBAAN D,FANG Y,et al.Genotypic and phenotypic characteristics of Dutch patients with early onset Parkinson's disease[J].Mov Disord,2009,24(2):196-203.
[9]ZHANG BR,HU ZX,YIN XZ,et al.Mutation analysis of parkin and PINK1 genes in early-onset Parkinson's disease in China [J].Neurosci Lett,2010,477(1):19-22.
[10]PAISAN-RUIZ C,BHATIA KP,LI A,et al.Characterization of PLA2G6 as a locus for dystonia-parkinsonism[J].Ann Neurol, 2009,65(1):19-23.
[11]WU Y,JIANG Y,GAO Z,et al.Clinical study and PLA2G6 mutation screening analysis in Chinese patients with infantile neuroaxonal dystrophy[J].Eur J Neurol,2009,16(2):240-245.
[12]SHI CH,TANG BS,WANG L,et al.PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort[J].Neurology,2011,77(1):75-81.
[13]康纪峰,唐北沙,郭纪锋.PLA2 G6基因与帕金森病[J].中华神经科杂志,2016(2):145-149.
[14]SEIRAFI M,KOZLOV G,GEHRING K.Parkin structure and function[J].FEBS J,2015,282(11):2076-2088.
[15]中华医学会神经病学分会帕金森病及运动障碍学组.中国帕金森病治疗指南 (第三版)[J].中华神经科杂志,2014(6): 428-433.
R742.5 (
2017-02-07)
A (责任编辑:李立)
10.3969/j.issn.1002-0152.2017.07.013
* 东南大学医学院神经精神研究所,东南大学附属中大医院神经内科(南京210009)
