脑胶质瘤生物学标记物和分子病理分型的研究进展
2017-09-03李子为蔡金全蒋传路
李子为,蔡金全,蒋传路
(哈尔滨医科大学,黑龙江哈尔滨150081)
脑胶质瘤生物学标记物和分子病理分型的研究进展
李子为,蔡金全,蒋传路
(哈尔滨医科大学,黑龙江哈尔滨150081)
脑胶质瘤作为最常见的颅内原发肿瘤,由于其诊断方式的局限性,造成患者预后较差.如何尽早精确诊断脑胶质瘤并制定相应的针对性治疗计划成为治疗胶质瘤的难题之一.随着临床研究的不断扩大和深入,生物学标记物及分子病理分型能够为多种疾病的诊断及治疗提供帮助.本文重点阐述脑胶质瘤生物学标记物及分子病理分型的研究进展,旨在为今后的胶质瘤临床诊断及治疗提供科学指导,改善患者预后,提高其生存质量.
胶质瘤;生物学标记物;分子病理分型
0 引言
脑胶质瘤是由于大脑和脊髓胶质细胞癌变所产生的、最常见的原发性颅脑肿瘤.常采用手术、放疗、化疗相结合的综合治疗方案对其进行治疗且预后较差.WHOⅣ级胶质母细胞瘤(glioblastoma,GBM)约占原发GBM的50%,其中位生存期仅14个月.
在WHO的分类中,尽管低级别(Ⅱ级)胶质瘤患者的预后及中位生存期较高级别(Ⅲ级、Ⅳ级)胶质瘤患者的预后更为有利,但50%~75%的低级别胶质瘤患者的肿瘤仍会继续生长和发展成为更高级别,最终导致患者的神经系统功能障碍和死亡.脑胶质瘤患者预后受肿瘤级别高低、病理类型不同、手术切除范围大小等多种因素影响.由于当前基础形态学对胶质瘤诊断的局限,对于胶质瘤病理类型、肿瘤生物学行为、患者预后和术后临床治疗无法提供太多帮助.
近年来研究发现胶质瘤生物学标记物及分子病理分型为精确诊断及制定治疗方案提供了重要的参考,本文就上述脑胶质瘤生物学标记物及分子病理分型的研究进展进行综述.
1 脑胶质瘤生物学标记物研究进展
1.1 异柠檬酸脱氢酶(isocitrate dehydrogenase,IDH)的突变在三羧酸循环中,IDH是一种限速酶,在细胞中主要通过催化异柠檬酸氧化脱羧生成α⁃酮戊二酸和还原型辅酶Ⅱ为细胞新陈代谢提供能量,同时α⁃酮戊二酸和还原型辅酶Ⅱ还可以作为生物合成的基本物质.IDH突变多发于青年患者,IDH1突变与脑胶质瘤的发生、发展及预后关系密切,各个级别脑胶质瘤患者中,IDH突变型意味着患者总生存期及无进展生存期更长.IDH1⁃R132H点突变占IDH总突变的约95%[1⁃2],在星形细胞瘤中发生比例为 60%~65%,原发性多形性胶质母细胞瘤仅占5%~20%,继发多形性胶质母细胞瘤约为80%,少突神经胶质瘤为75%~80%,少突星形细胞瘤为80%~85%[1,3⁃7].IDH基因突变被认为发生在脑胶质瘤发生的早期[3].IDH、TP53、ATRX基因突变是较低级别星形细胞瘤或继发性GBM的特征性分子标志物[8].
1.2 O6⁃甲基鸟嘌呤⁃DNA⁃甲基转移酶(O6⁃methyl⁃guanine DNA methyltransferase,MGMT)启动子区甲基化MGMT基因位于10q26,是一种编码修复O6⁃甲基鸟嘌呤的酶.在正常细胞中,MGMT启动子区域常处于低甲基化或非甲基化状态,当启动子区域甲基化或者高甲基化后,会导致染色质紧缩,抑制相应启动子结合进而导致MGMT转录水平下降抑制其表达.在肿瘤细胞中的MGMT蛋白可通过烷基化形成的O6位甲基化鸟嘌呤进行去甲基化,有效修复化疗药物造成的DNA损伤[9⁃11].研究表明,MGMT基因启动子甲基化的胶质母细胞瘤患者对化疗[12⁃14]更为敏感,生存期更长.因此,MGMT甲基化可作为主要预后标志物,还可以为化疗反应提供指导.
1.3 染色体1p和19q联合缺失染色体1p/19q联合缺失(loss of heterozygosity,LOH)在少突胶质细胞瘤中发生率为80%~95%[15],在少突星形细胞瘤中1p/19q联合缺失发生率约40%~60%[16].在星形细胞瘤中1p/19q缺失的低发生率,与ATRX和TP53基因突变并无相关联系[3,17],在少突胶质细胞瘤中,目前1p/19q联合缺失被认为是特征性分子标志物[18],并提示肿瘤患者对化疗药物敏感[19].目前认为在少突胶质细胞瘤患者中,1p/19q联合缺失的作用机制与FUBP1和CIC基因的突变有一定关系.
1.4 ATRX突变/缺失在大约80%的WHOⅡ级和Ⅲ级星形细胞瘤和继发胶质母细胞瘤中,α⁃地中海贫血/智力缺陷综合征X染色体连锁基因(α⁃thalas⁃semia/mental retardation syndrome X⁃linked,ATRX)核内表达缺失,而在WHOⅡ级和Ⅲ级原发胶质母细胞瘤和少突胶质细胞瘤中表达缺失率较低,这在鉴别肿瘤亚分型星形细胞瘤中发挥着重要作用[20⁃21].在星形细胞瘤患者中,大量研究[7]表明,低ATRX表达的患者预后更好.对于间变性胶质瘤患者的预后评估,ATRX突变、联合IDH突变及1p/19q联合缺失状态具有重要的参考价值[20,22⁃23].
1.5 端粒酶逆转录酶(telomerase reverse tran⁃scriptase,TERT)启动子区突变端粒酶的主要功能为延长端粒,维持细胞的增殖.近年来研究表明,特征性的TERT基因启动子区突变发生于胶质瘤细胞中,主要突变形式为C228T和C250T,发生的总频率约为55%,原发性胶质母细胞瘤的发生率约为55%~83%,少突胶质细胞瘤的发生率约为74%~78%.在突变的肿瘤组织中,TERT表达水平是野生型的6.1倍.在少突胶质细胞瘤样本中,98%的TERT启动子突变与1p/19q杂合性缺失相关联.在脑胶质瘤中,将TERT启动子突变与遗传学事件相结合,对于分子病理分型及预后判断有一定的参考价值[24⁃26].
1.6 表皮生长因子受体(epidermal growth factor receptor,EGFR)异常EGFR基因位于7号染色体p12区域,编码跨膜酪氨酸激酶受体.通过表皮生长因子(epidermal growth factor,EGF)等配体的结合可以促进其自身的磷酸化,磷酸化的EGFR可以影响多个信号通路的活性,进而改变细胞生物学行为,如在星型胶质细胞瘤和多形胶质母细胞瘤中,PTEN基因(gene of phosphate and tension homology detected on chromsome ten,PTEN)突变联合EGFR扩增的患者预后较差[27].在高级别胶质瘤中最常见的EGFR突变类型是EGFR的扩增和外显子2~7缺失(EGFRvⅢ型突变),在癌症基因组图谱计划(the cancer ge⁃nome atlas,TCGA)的经典型[28]及Philips分型中的增殖型和间质亚型中,EGFR扩增的发生率为94%[29-30].近年研究发现,EGFR40%的扩增率是原发性胶质母细胞瘤中最高的[31-32].
1.7 PTPRZ1⁃MET融合基因2014年,中国脑胶质瘤基因组图谱计划(CGGA)发布了中国人脑胶质瘤融合基因全景图.该研究提供了首个针对中国人群脑胶质瘤的高通量第二代测序技术(next generation se⁃quencing,NGS),揭示了脑胶质瘤融合基因全景图,发现了新的脑胶质瘤分子特征,首次在继发胶质母细胞瘤中发现了重复出现的PTPRZ1⁃MET融合基因,并描述了其发生融合的四种方式.这一研究为脑胶质瘤的临床诊断和个性化治疗提供了分子基础(图1).
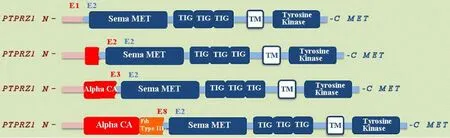
图1 PTPRZ1、MET及PTPRZ1⁃MET融合蛋白示意图[51]
1.8 Ki⁃67Ki⁃67因其在活细胞中表达的特异性可作为指标用于评定细胞增殖能力.Preusser等[33]在大规模临床研究中发现Ki⁃67是一个非常重要的单因素分析预后指标.Ki⁃67和磷酸化组蛋白H3(phos⁃pho⁃histone H3,PHH3)在弥漫性脑胶质瘤中有评估患者预后的价值[34-35].但是相比Ki⁃67,IDH突变状态和MGMT基因启动子甲基化状态则是更为重要的预后因素.
2 脑胶质瘤分子病理分型研究进展
2.1 转录组学分型胶质母细胞瘤可根据基因转录表达分为4个亚型:经典型(classical)、神经元型(neural)、间质性(mesenchymal)和前神经元型(pro⁃neural)[30,36](图2).四种亚型中,原发性胶质母细胞瘤classical型以7号染色体扩增和10号染色体缺失为特征,可见于EGFR扩增和突变.在25%的原发性胶质母细胞瘤中可见EGFRvⅢ型突变[37].EGFR点突变约占肿瘤的25%,但该亚型未见有肿瘤抑制基因TP53的突变(图3).而血小板源性生长因子受体(platelet derived growth factor receptor,PDGFR)扩增与前神经元型原发性胶质母细胞瘤有关,包括4号染色体q12上的KDR和KIT基因突变,和少突胶质细胞发育基因Olig的过表达[30].
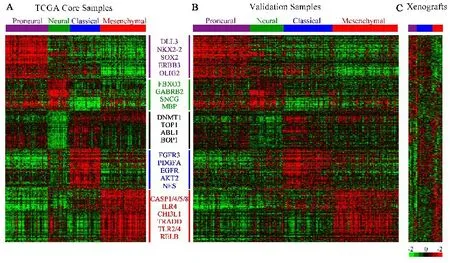
图2 TCGA根据转录水平的差异将胶质母细胞瘤分为四个亚型

图3 在164个胶质母细胞瘤中,转录组测序确定EGFR转录本变异率
IDH突变可见于前神经元型原发性GBM,与总生存期增加有关[38⁃40].通过Consensus Clustering聚类方法,应用中国人脑胶质瘤基因组学数据库样本的全基因组表达谱芯片数据,根据IDH突变水平和患者年龄及预后提出了中国人群中脑胶质瘤的三分子亚型:G1型,G2型和G3型[41].G1亚型中,患者年轻、预后好,IDH突变频率极高.G3亚型中,患者年龄大、预后差,IDH突变率低.G2亚型中,患者年龄分布,预后以及IDH突变率介于G1和G3亚型之间.与G1和G3亚型相比,G2亚型除了IDH突变水平的不同,1p/19q杂合性缺失频率也较高.这种新的分型方法可以更准确地反映中国人群脑胶质瘤流行病学和分子遗传学特征,为精准地制定患者个体诊疗计划提供理论依据(图4).
2014年,北京师范大学生命科学学院樊小龙教授团队提出了基于分子共表达网络的脑胶质瘤分子分型系统.该研究团队利用EGFR、PDGFRA在神经发生过程和脑胶质瘤中发挥重要作用的两个酪氨酸激酶受体分子建立了脑胶质瘤中EGFR和PDGFRA共表达的分子网络,可将脑胶质瘤分为EM、PM、EM⁃low+PMlow三大类型[42],代表着两个特征性的调控通路.相比EM型,PM型和PMlow+EMlow型预后较好.该分型是对于目前基于形态学的脑胶质瘤临床诊断分类的重要补充,为脑胶质瘤的靶向治疗和特异性分子标记物筛选提供了一定的参考价值(图5).

图4 中国人群脑胶质瘤三分型

图5 EM/PM分子分型及其与胶质瘤患者预后之间的关系
2.2 表观遗传组学分型脑胶质瘤可以根据基因组特定区域在表观遗传修饰分为CpG岛甲基化表型(glioma⁃CpG island methylator phenotype,G⁃CIMP)和非G⁃CIMP型[43⁃44](图6).
相较于非甲基化型,G⁃CIMP亚型的脑胶质瘤患者更为年轻,预后相对较好.MGMT甲基化是G⁃CI⁃MP亚型患者经过治疗后反应的一种生物标记物.同时,IDH突变的脑胶质瘤在某些G⁃CIMP存在超甲基化,而多数G⁃CIMP脑胶质瘤存在IDH突变.由此可见,G⁃CIMP是WHOⅣ级胶质瘤较为理想的预后标志物,IDH野生型或非G⁃CIMP的WHOⅣ级胶质瘤,其恶性程度高,预后较差[30,45].

图6 胶质母细胞瘤启动子区甲基化状态的聚类分析
2012年,德国研究团队应用Illumina 450K甲基化芯片对151个成人胶质母细胞瘤和59个儿童胶质母细胞瘤样本进行甲基化分析,并进一步将TCGA甲基化分型细化为K27型、G34型、RTKⅠ‘PDGFRA’型和间质型,与IDH型(TCGA,G⁃CIMP型)和RTKⅡ‘classic’型(TCGA,Cluster 2型),构成六分型系统[46].H3F3A基因的突变可以通过改变组蛋白H3.3、K27和G34位点甲基化水平,从而在表观遗传学上调控基因转录,进而改变基因表达水平,最终影响肿瘤的发生发展(图7).
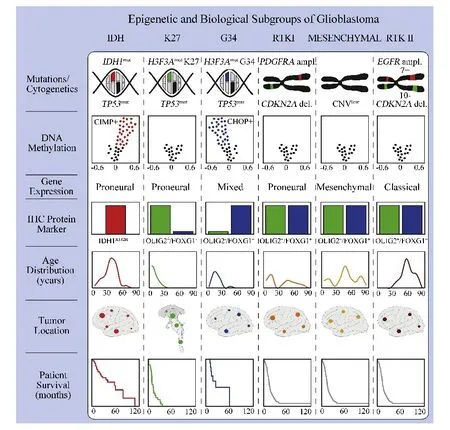
图7 胶质母细胞瘤甲基化分型示意图
2.3 影像组学分型随着影像分析技术的进步,对胶质瘤影像学特征进行定量化分析已经逐步成为现实.根据胶质母细胞瘤基因表达和影像学信号差异的关系,可建立依据定量影像特征联合基因蛋白表达预测肿瘤患者预后的评估模型.2015年斯坦福大学Gevaert教授收集265例胶质母细胞瘤患者的MRI影像学数据,采用定量成像管道,结合灰度值,纹理,病变边界清晰度等特征,富集产生388个图像特征,代表病变的单层和多层二维特征[47].
根据患者定量影像特征的Consensus聚类分析,确定“三分组”方案在发现组和验证组产生最大k值和最小AUC面积增量的同时还能最大化组内一致性,最小化重复后歧义发生率.在发现组,“一类分组”包含36例患者,“二类分组”包含51例患者,“三类分组”包含34例患者.验证组中,cluster1包含25例患者,cluster2包含107例患者,cluster3包含12例患者.利用基因芯片显著性分析,每个特征代表肿瘤像素强度及其特征.“一类分组”被肿瘤高度不规则特征定义,称为“pre⁃multifocal GBM cluster”(前多灶型GBM).“二类分组”被带有规则边缘肿瘤特征定义,称为“spherical GBM cluster”(球型GBM).“三类分组”特征性表现为中央低信号,边缘高信号,称为“rim⁃enhancing GBM cluster”(边缘增强型GBM)(图8).预后分析显示,前多灶型GBM患者预后最差,球型GBM患者预后一般,边缘增强型GBM患者预后最佳.TCGA四分型、IDH突变等遗传学变化在3个影像分组间并无差异.综合TCGA表达谱和CNV谱发现前多灶型GBM中c⁃Kit干细胞因子受体通路表达上调,球型GBM中21个通路下调,包括c⁃Kit,VEG⁃FR,PDGFRA,FOXA等.而边缘增强型GBM表现为包括WNT、PDGFRB、VEGFR等信号通路在内的31条通路上调.

图8 MRI特征定义胶质母细胞瘤亚型及其与预后、信号通路之间的关系
2.4 经典分子标记物分型基于脑胶质瘤遗传学事件的研究进展,人们对脑胶质瘤关键基因突变有了更深的认识.
在IDH,ATRX,CIC等胶质瘤病理类型特征性突变的基础上,2012年杜克大学病理科教授Hai Yan教授和约翰霍普金斯医学院Luis Diaz教授对363个胶质母细胞瘤样本进行突变位点分析,构建IDH1/ATRX(Ⅰ⁃A),IDH1/CIC/FUBP1(Ⅰ⁃CF)和Ⅰ⁃X三种亚型.其中,Ⅰ⁃A亚型包括IDH和ATRX突变,Ⅰ⁃CF亚型包括IDH突变和CIC突变或FUBP1突变或1p/19q缺失.正常脑组织细胞向胶质瘤发展过程中的早期多为Ⅰ⁃A和Ⅰ⁃CF亚型.Ⅰ⁃CF亚型特征性的表现为WHOⅡ/Ⅲ少突胶质细胞瘤,患者一般拥有最理想的总生存期,为96个月.Ⅰ⁃A特征性的表现为WHOⅡ/Ⅲ星形细胞瘤或WHOⅣ级继发胶质母细胞瘤,患者中位生存期为51个月.上述两种亚型阴性的肿瘤称谓Ⅰ⁃X亚型,患者预后最差,生存期为13个月(图 9)[48].
2013年德国癌症研究中心(DKFZ)在国际顶级神经病理杂志Acta Neuropathologica发表的论文显示在间变性脑胶质瘤中,ATRX表达缺失和1p/19q联合缺失对患者预后的判断及相关性明显优于传统病理分型(图10).此研究将未发生1p/19q联合缺失的混合胶质瘤及间变星形胶质瘤统称为“分子星型胶质瘤”(无论是否伴随ATRX突变).IDH野生型间变胶质瘤被认为是“分子胶质母细胞瘤”.并根据患者预后的良恶,由良至差依次为:分子少突胶质细胞瘤、分子星形细胞瘤、分子胶质母细胞瘤.传统混合胶质瘤的诊断局限于病理医生的主观性,容易导致人为误差,而ATRX联合1p/19q缺失状态可以在分子水平帮助诊断各型混合胶质瘤.另外,此次研究中发现,在IDH突变的前提下,ATRX表达缺失的星形细胞瘤患者预后好[49].根据分子标记物的变异状态和患者的临床特点,2015年Mayo研究所USFD团队对1087例胶质瘤的IDH、1p/19q和TERT启动子区状态进行检测,将患者分为5个分子亚型.同时,通过11590例对照组患者用来评估胶质瘤患者的生殖系变异.结果显示,在615例WHOⅡ/Ⅲ级胶质瘤中,29%的患者同时带有三个变异,称为“三阳性”胶质瘤,7%的患者三个变异均为阴性,我们称之为“三阴性”胶质瘤,10%的患者仅带有TERT启动子区突变,5%的患者带有IDH和TERT启动子区突变,45%的患者仅带有IDH突变,另外5%的患者呈其它组合.在472例胶质母细胞瘤患者中,不到1%的患者呈“三阳性”,2%的患者有IDH和TERT启动子区突变,7%的患者仅有IDH突变,17%的患者为“三阴性”胶质瘤,74%的患者仅有TERT启动子区突变.仅带有IDH突变的胶质瘤患者诊断时年龄最小,为37岁,仅含有TERT启动子区突变的胶质瘤患者诊断时年龄最大,为59岁.这种分子分型在低级别(Ⅱ~Ⅲ级)胶质瘤患者中可以独立评估预后,但在胶质母细胞(Ⅳ级)瘤患者中不能独立评估预后.并且与生殖系突变相关.CCDC26 SNP(rs55705857)与IDH突变的胶质瘤有关,PHLDB1 SNP(rs498872)与仅带有IDH突变的胶质瘤有关,TERC(rs1920116),TERT(rs2736100)和RTEL1(rs6010620)SNPs是仅带有TERT启动子区突变的保护性因素(图11)[50].
2.5多组学联合分型弥散低级别胶质瘤和中级别(intermediate⁃grade)胶质瘤具有高度多样的临床特征.其中一些肿瘤较为稳定,而另一些则可迅速进展到胶质母细胞瘤.这种不确定性部分原因是由于组织病理诊断的差异性造成的.

图10 根据形态学病理和分子标记物的间变性胶质瘤分子分型
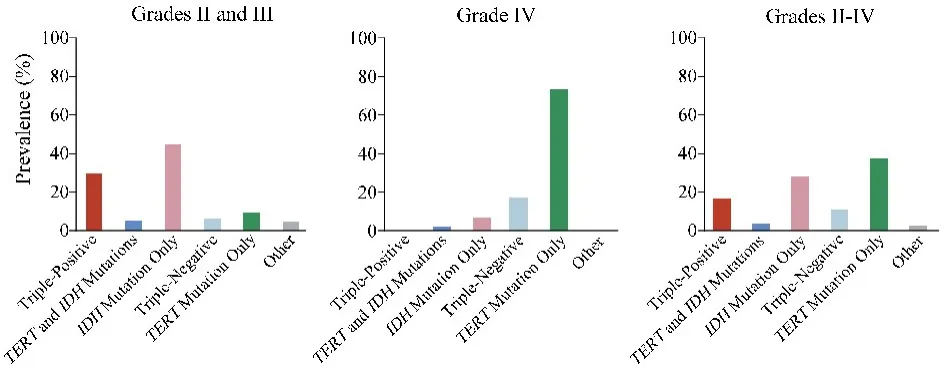
图11 基于IDH、TERT、1p/19q状态的分子分型在各个级别中的比例分布
较低级别脑胶质瘤中的常见变异,如 IDH、TP53、ATRX突变和染色体1p/19q联合缺失状态已经用作临床胶质瘤的诊断性标记物.美国TCGA团队对293例成人较低级别胶质瘤进行了全基因组分析,包括外显子测序、DNA拷贝数变化、DNA甲基化谱、信使RNA表达谱,microRNA表达谱和特异蛋白表达谱.这些数据被用于整合分析其相关性和临床意义.研究结果显示,突变、RNA,DNA拷贝数,DNA甲基化等数据的非监督聚类揭示了较低级别胶质瘤中三个协调、无重复且与患者预后密切相关的亚分层(图12).这种亚分层更易通过IDH、1p/9q、TP53等变异状态来定义,而不是以传统组织病理学来分类.带有IDH突变和1p/9q联合缺失的较低级别胶质瘤患者预后最好,这类肿瘤一般带有CIC、FUBP1、NOTCH1和TERT启动子区突变.而IDH突变且没有1p/9q联合缺失的较低级别胶质瘤一般均带有TP53突变和ATRX突变.IDH野生型的较低级别胶质瘤具有与原发胶质母细胞瘤类似的基因组学变异和临床特征(图13).
3 讨论
随着临床研究及科学技术的进步,生物学标记物和分子病理分型逐渐广泛应用于多种疾病的诊断,其在脑胶质瘤诊断及治疗方案制定中也发挥着重要价值,成为精确诊断及治疗的手段之一.生物学标记物及分子病理分型能够为尽早诊断及治疗脑胶质瘤提供重要参考,以延长患者中位生存期,改善预后,提高其生活质量.
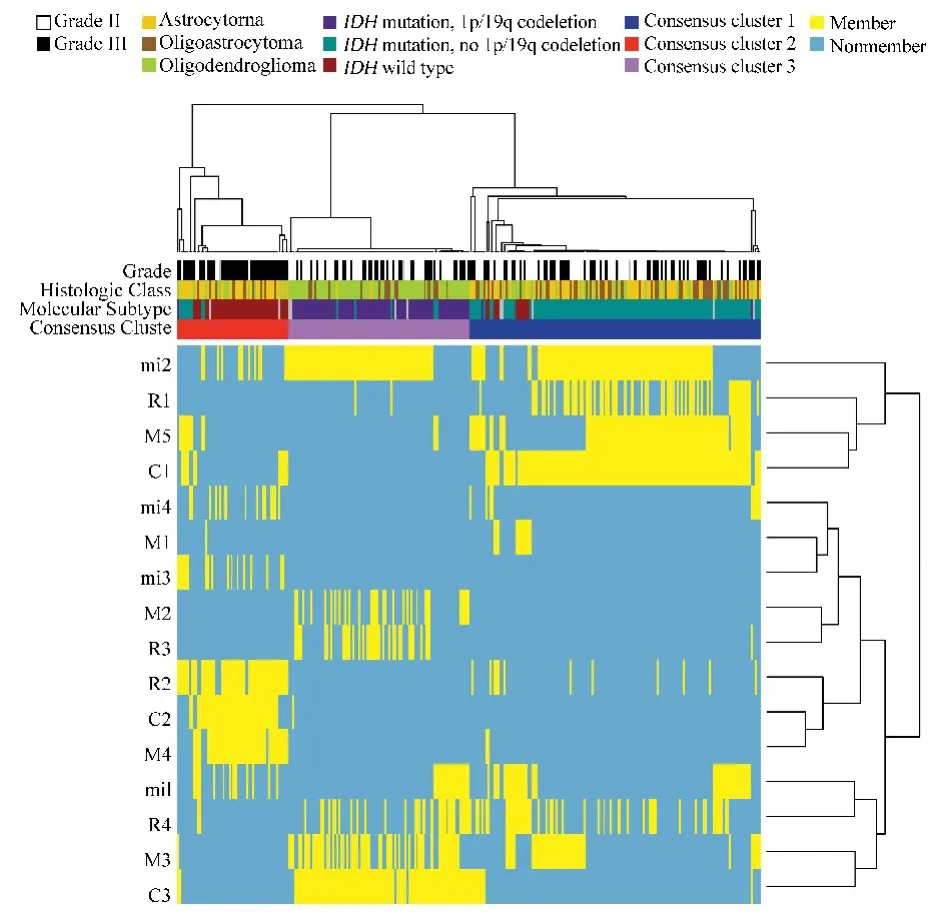
图12 Cluster of clusters分析显示在较低级别胶质瘤中存在三个分子亚型
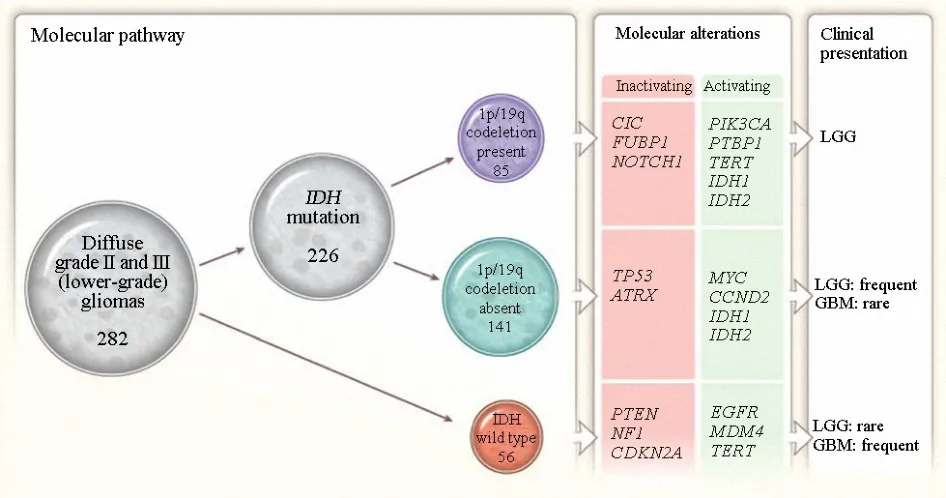
图13 三分型的基因组学变化和临床特征总览
[1]Horbinski C,Kofler J,Kelly LM,et al.Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin⁃fixed,paraf⁃fin⁃embedded glioma tissues[J].J Neuropathol Exp Neurol,2009,68(12):1319-1325.
[2]Capper D,Reuss D,Schittenhelm J,et al.Mutation⁃specific IDH1 antibody differentiates oligodendrogliomas and oligoastrocytomas from other brain tumors with oligodendroglioma⁃like morphology[J].Acta Neuropathol,2011,121(2):241-252.
[3]Watanabe T,Nobusawa S,Kleihues P,et al.IDH1 mutations are early events in the development of astrocytomas and oligodendroglio⁃mas[J].Am J Pathol,2009,174(4):1149-1153.
[4]Hartmann C,Meyer J,Balss J,et al.Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas[J].Acta Neuropathol,2009,118(4):469-474.
[5]Balss J,Meyer J,Mueller W,et al.Analysis of the IDH1 codon 132 mutation in brain tumors[J].Acta Neuropathol,2008,116(6):597-602.
[6]Ichimura K,Pearson DM,Kocialkowski S,et al.IDH1 mutations are present in the majority of common adult gliomas but rare in pri⁃mary glioblastomas[J].Neuro Oncol,2009,11(4):341-347.
[7]Cai J,Yang P,Zhang C,et al.ATRX mRNA expression combinedwith IDH1/2 mutational status and Ki⁃67 expression refines the molecular classification of astrocytic tumors:evidence from the whole transcriptome sequencing of 169 samples samples[J].Oncotarget,2014,5(9):2551-2561.
[8]Nobusawa S,Watanabe T,Kleihues P,et al.IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas[J].Clin Cancer Res,2009,15(19):6002-6007.
[9]Bleeker FE,Molenaar RJ,Leenstra S.Recent advances in the molecular understanding of glioblastoma[J].J Neurooncol,2012,108(1):11-27.
[10]Weller M,Stupp R,Reifenberger G,et al.MGMT promoter methyl⁃ation in malignant gliomas:ready for personalized medicine[J].Nat Rev Neurol,2010,6(1):39-51.
[11]Kaina B,Christmann M,Naumann S,et al.MGMT:key node in the battle against genotoxicity,carcinogenicity and apoptosis induced by alkylating agents[J].DNA Repair(Amst),2007,6(8):1079-1099.
[12]van den Bent MJ,Dubbink HJ,Sanson M,et al.MGMT promoter methylation is prognostic but not predictive for outcome to adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors:a report from EORTC Brain Tumor Group Study 26951[J].J Clin Oncol,2009,27(35):5881-5886.
[13]Everhard S,Kaloshi G,Criniere E,et al.MGMT methylation:a marker of response to temozolomide in low⁃grade gliomas[J].Ann Neurol,2006,60(6):740-743.
[14]Wick W,Hartmann C,Engel C,et al.NOA⁃04 randomized phaseⅢ trial of sequential radiochemotherapy of anaplastic glioma with procarbazine,lomustine,and vincristine or temozolomide[J].J Clin Oncol,2009,27(35):5874-5880.
[15]Reifenberger G,Louis DN.Oligodendroglioma:toward molecular definitions in diagnostic neuro⁃oncology[J].J Neuropathol Exp Neurol,2003,62(2):111-126.
[16]Lassman AB,Iwamoto FM,Cloughesy TF,et al.International retro⁃spective study of over 1000 adults with anaplastic oligodendroglial tumors[J].Neuro Oncol,2011,13(6):649-659.
[17]Metellus P,Coulibaly B,Colin C,et al.Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO gradeⅡgliomas with dismal prognosis[J].Acta Neuropathol,2010,120(6):719-729.
[18]Aldape K,Burger PC,Perry A.Clinicopathologic aspects of 1p/19q loss and the diagnosis of oligodendroglioma[J].Arch Pathol Lab Med,2007,131(2):242-251.
[19]Kaloshi G,Benouaich⁃Amiel A,Diakite F,et al.Temozolomide for low⁃grade gliomas:predictive impact of 1p/19q loss on response and outcome[J].Neurology,2007,68(21):1831-1836.
[20] Haberler C,Wohrer A.Clinical Neuropathology practice news 2⁃2014:ATRX,a new candidate biomarker in glioma[J].Clin Neu⁃ropathol,2014,33(2):108-111.
[21]Kannan K,Inagaki A,Silber J,et al.Whole⁃exome sequencing identifies ATRX mutation as a key molecular determinant in lower⁃grade glioma[J].Oncotarget,2012,3(10):1194-1203.
[22]Reuss DE,Sahm F,Schrimpf D,et al.ATRX and IDH1⁃R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an“integrated”diagnostic approach for adult astrocytoma,oligodendroglioma and glioblastoma[J].Acta neuropathol,2015,129(1):133-146.
[23]Leeper HE,Caron AA,Decker PA,et al.IDH mutation,1p19q codeletion and ATRX loss in WHO gradeⅡgliomas[J].Oncotarget,2015,6(30):30295-30305.
[24]Arita H,Narita Y,Fukushima S,et al.Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss[J].Acta Neuropathol,2013,126(2):267-276.
[25]Killela PJ,Reitman ZJ,Jiao Y,et al.TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self⁃renewal[J].Proc Natl Acad Sci U S A,2013,110(15):6021-6026.
[26]Killela PJ,Pirozzi CJ,Healy P.Mutations in IDH1,IDH2,and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas[J].Oncotarget,2014,5(6):1515-1525.
[27]Smith JS,Tachibana I,Passe SM,et al.PTEN mutation,EGFR amplification,and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme[J].J Natl Cancer Inst,2001,93(16):1246-1256.
[28]Verhaak RG,Hoadley KA,Purdom E,et al.Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma charac⁃terized by abnormalities in PDGFRA,IDH1,EGFR,and NF1[J].Cancer Cell,2010,17(1):98-110.
[29]Phillips HS,Kharbanda S,Chen R,et al.Molecular subclasses of high⁃grade glioma predict prognosis,delineate a pattern of disease progression,and resemble stages in neurogenesis[J].Cancer Cell,2006,9(3):157-173.
[30]Gao G,Ren S,Li A,et al.Epidermal growth factor receptor⁃tyrosine kinase inhibitor therapy is effective as first⁃line treatment of advanced non⁃small⁃cell lung cancer with mutated EGFR:A meta⁃analysis from six phaseⅢ randomized controlled trials[J].Int J Cancer,2012,131(5):E822-829.
[31]Ohgaki H,Kleihues P.Genetic pathways to primary and secondary glioblastoma[J].Am J Pathol,2007,170(5):1445-1453.
[32]Ohgaki H,Dessen P,Jourde B,et al.Genetic pathways to glioblastoma:a population⁃based study[J].Cancer Res,2004,64(19):6892-6899.
[33]Preusser M,Hoeftberger R,Woehrer A,et al.Prognostic value of Ki67 index in anaplastic oligodendroglial tumours⁃⁃a translational study of the European Organization for Research and Treatment of Cancer Brain Tumor Group[J].Histopathology,2012,60(6):885-894.
[34]Habberstad AH,Gulati S,Torp SH.Evaluation of the proliferation markers Ki⁃67/MIB⁃1,mitosin,survivin,pHH3,and DNA topoi⁃somerase IIɑ in human anaplastic astrocytomas⁃⁃an immunohisto⁃chemical study[J].Diagn Pathol,2011,6:43.
[35]Colman H,Giannini C,Huang L,et al.Assessment and prognostic significance of mitotic index using the mitosis marker phospho⁃his⁃tone H3 in low and intermediate⁃grade infiltrating astrocytomas[J].Am J Surg Pathol,2006,30(5):657-664.
[36]Lai A,Kharbanda S,Pope WB,et al.Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin[J].J Clin Oncol,2011,29(34):4482-4490.
[37]Kastenhuber ER,Huse JT,Berman SH,et al.Quantitative assess⁃ment of intragenic receptor tyrosine kinase deletions in primary glioblastomas:their prevalence and molecular correlates[J].Acta neuropathol,2014,127(5):747-759.
[38]Parsons DW,Jones S,Zhang X,et al.An integrated genomic analysis of human glioblastoma multiforme[J].Science,2008,321(5897):1807-1812.
[39]Yan H,Parsons DW,Jin G,et al.IDH1 and IDH2 mutations in gliomas[J].N Engl J Med,2009,360(8):765-773.
[40]Labussière M,Idbaih A,Wang XW,et al.All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2[J].Neurology,2010,74(23):1886-1890.
[41]Yan W,Zhang W,You G,et al.Molecular classification of gliomas based on whole genome gene expression:a systematic report of 225 samples from the Chinese Glioma Cooperative Group[J].Neuro Oncol,2012,14(12):1432-1440.
[42]Sun Y,Zhang W,Chen D,et al.A glioma classification scheme based on coexpression modules of EGFR and PDGFRA[J].Proc Natl Acad Sci U S A,2014,111(9):3538-3543.
[43]Noushmehr H,Weisenberger DJ,Diefes K,et al.Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma[J].Cancer Cell,2010,17(5):510-522.
[44]Turcan S,Rohle D,Goenka A,et al.IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype[J].Nature,2012,483(7390):479-483.
[45]Brennan CW,Verhaak RG,McKenna A,et al.The somatic genomic landscape of glioblastoma[J].Cell,2013,155(2):462-477.
[46]Sturm D,Witt H,Hovestadt V,et al.Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma[J].Cancer Cell,2012,22(4):425-437.
[47]Itakura H,Achrol AS,Mitchell LA,et al.Magnetic resonance image features identifyglioblastoma phenotypic subtypeswith distinct molecular pathway activities[J].Sci Transl Med,2015,7(303):303ra138.
[48]Jiao Y,Killela PJ,Reitman ZJ,et al.Frequent ATRX,CIC,FUBP1 and IDH1 mutations refine the classification of malignant gliomas[J].Oncotarget,2012,3(7):709-722.
[49]Wiestler B,Capper D,Holland⁃Letz T,et al.ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis[J].Acta Neuropathol,2013,126(3):443-451.
[50]Eckel⁃Passow JE,Lachance DH,Molinaro AM,et al.Glioma Groups Based on 1p/19q,IDH,and TERT Promoter Mutations in Tumors[J].N Engl J Med,2015,372(26):2499-2508.
[51]Bao ZS,Chen HM,Yang MY,et al.RNA⁃seq of 272 gliomas revealed a novel,recurrent PTPRZ1⁃MET fusion transcript in second⁃ary glioblastomas[J].Genome Res,2014,24(11):1765-1773.
Research advances on the biological markers and classification of molecular pathology of glioma
LI Zi⁃Wei,CAI Jin⁃Quan,JIANG Chuan⁃Lu
Harbin Medical University,Harbin 150081,China
Brain glioma is the most common intracranial primary tumor,due to the limitations of its diagnostic methods,resulting in poor prognosis of patients.How to diagnose brain gliomas as soon as possible and develop appropriate targeted treatment plans become one of the problems in the treatment of gliomas.With development of clinical research and scientific advances,biology markers and molecular pathology have been found to be helpful in the diagnosis and treatment of various diseases.Therefore,this paper focuses on research progress of biomechanical markers and molecular pathology of glioma,which are aimed at providing sci⁃entific guidance for the future of glioma clinical diagnosis and treatment,and improve the prognosis and life quality of patients.
glioma;biological markers;molecular pathology
R739.41
A
2095⁃6894(2017)07⁃21⁃08
2017-04-25;接受日期:2017-05-12
中国抗癌协会神经肿瘤专业委员会神经肿瘤研究基金(CSNO⁃2016⁃MSD12);哈尔滨医科大学创新科学研究资助项目(2017LCZX37)
李子为.硕士生.E⁃mail:765310397@qq.com
蒋传路.博士,教授,主任医师.研究方向:神经外科.E⁃mail:15124572707@126.com
