Gut microbiota dysbiosis in patients with nonalcoholic fatty liver disease
2017-08-16FengShenRuiDanZhengXingQiangSunWenJinDingXiaoYingWangandJianGaoFan
Feng Shen, Rui-Dan Zheng, Xing-Qiang Sun, Wen-Jin Ding, Xiao-Ying Wang and Jian-Gao Fan
Shanghai, China
Gut microbiota dysbiosis in patients with nonalcoholic fatty liver disease
Feng Shen, Rui-Dan Zheng, Xing-Qiang Sun, Wen-Jin Ding, Xiao-Ying Wang and Jian-Gao Fan
Shanghai, China
BACKGROUND:Gut microbiota plays a signif i cant role in the pathogenesis of non-alcoholic fatty liver disease (NAFLD). This study aimed to assess the contribution of gut microbiota dysbiosis to the pathogenesis of NAFLD.
METHODS:Forty-seven human feces samples (25 NAFLD patients and 22 healthy subjects) were collected and 16S rDNA amplicon sequencing was conducted on Hiseq 2000 platform. Discrepancy of species composition between controls and NAFLD group was def i ned by Metastats analysis underPvalue<0.01.
RESULTS:NAFLD patients harbored lower gut microbiota diversity than healthy subjects did. In comparison to the control group, the Proteobacteria (13.50%) and Fusobacteria (2.76%) phyla were more abundant in NAFLD patients. Additionally, the Lachnospiraceae (21.90%), Enterobacteriaceae (12.02%), Erysipelotrichaceae (3.83%), and Streptococcaceae (1.39%) families, as well as theEscherichia_Shigella(10.84%),Lachnospiraceae_Incertae_Sedis(7.79%), andBlautia(4.95%) genera were enriched in the NAFLD group. However, there was a lower abundance ofPrevotellain the NAFLD group than that in the control group (5.83% vs 27.56%,P<0.01). The phylum Bacteroidetes (44.63%) also tended to be more abundant in healthy subjects, and the families Prevotellaceae (28.66%) and Ruminococcaceae (26.44%) followed the same trend. Compared to those without non-alcoholic steatohepatitis (NASH), patients with NASH had higher abundance of genusBlautia(5.82% vs 2.25%;P=0.01) and the corresponding Lachnospiraceae family (24.33% vs 14.21%;P<0.01). Patients with signif i cant fi brosis had a higher abundance of genusEscherichia_Shigella(12.53% vs 1.97%;P<0.01) and the corresponding Enterobacteriaceae family (13.92% vs 2.07%;P<0.01) compared to those with F0/F1 fi brosis.
CONCLUSIONS:NAFLD patients and healthy subjects harbor varying gut microbiota. In contrast to the results of previous research on children, decreased levels ofPrevotellamight be detrimental for adults with NAFLD. The increased level of the genusBlautia, the family Lachnospiraceae, the genusEscherichia_Shigella, and the family Enterobacteriaceae may be a primary contributor to NAFLD progression.
(Hepatobiliary Pancreat Dis Int 2017;16:375-381)
gut microbiota; fatty liver disease; non-alcoholic steatohepatitis; fi brosis
Introduction
Non-alcoholic fatty liver disease (NAFLD) is currently the most prevalent chronic liver disease in industrialized countries, affecting nearly 20%-30% of the population.[1]The spectrum of NAFLD ranges from simple steatosis to non-alcoholic steatohepatitis (NASH), fi brosis, cirrhosis, and even end-stage liver disease. However, the pathogenesis of NAFLD/NASH is still not well def i ned. Recently, many studies have implicated gut microbiota (GM) as a leading cause of NAFLD/NASH.[2,3]The impact of GM on NAFLD/NASH has been attributed to deterioration of intestinal barrier functionality,[4]increased gut permeability,[5]intestinal endotoxemia,[6]endogenous alcohol,[7]up-regulation of hepaticde novolipogenesis and triglyceride synthesis,[8]reduction of choline bioavailability,[9]and aggravation of insulin resistance.[10]All of these studies have demonstrated that GM may play a very important role in thepathogenesis and progression of simple fatty liver, NASH, fi brosis, and cirrhosis.[11]
However, the abundance of fecal bacterial species was signif i cantly variable between different subjects,[12]and the GM structure in NAFLD patients was far from clear. To understand whether and how GM contributes to NAFLD pathogenesis and progression, we collected fecal samples from 47 subjects who did not consume alcohol (25 NAFLD patients and 22 healthy subjects) and performed 16S rDNA amplicon analysis for each sample. In addition to providing insight into the contribution of GM to NAFLD, this study will be a valuable reference to better understand the prevention and treatment of NAFLD.
Methods
Study population
Consecutive patients (aged ≥18 years) who underwent liver biopsies and met the diagnostic criteria of NAFLD[13]were prospectively recruited between May 2013 and July 2013 from Xinhua Hospital, Shanghai Jiaotong University. We excluded patients with secondary causes of hepatic steatosis (such as the use of systemic corticosteroids or total parenteral nutrition), infection with the hepatitis B or C virus, autoimmune liver disorders, or decompensated cirrhosis. In addition, we included healthy subjects who had no clinical signs or symptoms of illness, including evidence of viral hepatitis and metabolic syndromes, such as diabetes mellitus and high serum cholesterol. For healthy subjects, we used Fibroscan to rule out liver fi brosis and hepatic steatosis. The liver stiffness measurement (LSM) <5.8 kPa[14]and controlled attenuation parameter (CAP) <215 dB/m[15]were used to evaluate whether the healthy subjects experienced signif i cant fi brosis or hepatic steatosis. All subjects (both NAFLD patients and controls) with a history of excessive alcohol consumption (140 g per week for men, 70 g for women) within the past 12 months, “abnormal” dietary habits (e.g., vegetarian diet), and current or previous use of antibiotics (within the last 3 months) were also excluded. This study was approved by the Ethics Committees of Xinhua Hospital. After providing an adequate explanation, consent for participation in this study was provided by all subjects prior to sample collection.
Clinical and laboratory assessment
Demographic information was collected, including age, gender, history of drinking, hypertension, and type 2 diabetes mellitus. Anthropometric measurements were also recorded, such as BMI (kg/m2), waist circumference (cm), and hip circumference (cm). Laboratory tests were performed within one week of liver biopsy. Commercially available enzyme immunoassays were used to determine the serum HBsAg and anti-HCV levels. Serum fasting plasma glucose, alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin, total bilirubin, total cholesterol, triglycerides, high-density lipoprotein (HDL), and low-density lipoprotein (LDL) levels were measured using a standard testing kit or automatic system.
Liver histology
For the NAFLD group, a percutaneous liver biopsy was performed using an 18-gauge BARD Max-Core Disposable Core Biopsy Instrument (BARD Biopsy Systems, Tempe, AZ, USA) at the right lobe, under real-time ultrasound guidance. All samples ≥16 mm were included in at least six portal tracts. The liver biopsy specimens were fi xed in formalin, embedded in paraff i n, and stained with HE, Masson’s trichrome, and reticulin. Liver histology was assessed by an experienced hepatopathologist (Wang XY) who was blind to the clinical data. The proposed NAFLD activity score (NAS) was the unweighted sum of the scores for steatosis (0-3), lobular inf l ammation (0-3), and hepatocellular ballooning (0-2), and a score ≥5 was indicative of NASH.[16]Fibrosis of NAFLD was staged as follows: F0, absence of fi brosis; F1, perisinusoidal or periportal fi brosis; F2, perisinusoidal and portal/periportal fi brosis; F3, septal or bridging fi brosis; and F4, cirrhosis. Fibrosis ≥2 was considered as signif i cant fi brosis. Liver steatosis was categorized by visual assessment, as S1 (5%-33%), S2 (34%-66%), or S3 (>66%).
Fecal sample collection
Stool samples were obtained at the homes of each participant and were immediately frozen in their home freezer. Frozen samples were stored within insulating polystyrene foam containers, and stored at -80 ℃ until analysis. The time span from sampling to delivery to the Xinhua Hospital was intended to be as short as possible with a maximum of 24 hours.
DNA extraction, PCR amplif i cation, and sequencing
A frozen aliquot (200 mg) of each fecal sample was suspended in 250 μL of 4 mol/L guanidine thiocyanate, 0.1 mol/L Tris (pH 7.5), and 40 μL of 10% N-lauroyl sarcosine. Then, DNA extraction was conducted using the bead beating method, and DNA quality control was performed using a Qubit fl uorometer. PCR was performed to produce V3/V5 hypervariable regions of the 16S rDNA gene using the conserved primers 926F (5’-CCG TCA ATT CMT TTG AGT TT-3’) and 338R (5’-ACT CCT ACG GGA GGC AGC AG-3’). PCR fragments weresequenced with the 454 GS-FLX platform (Roche) under standard instruction.
Statistical analysis
The multiplexed samples were analyzed in Mothur (Version 1.31.2, http://www.mothur.org/)[17]based on the presence of the unique barcodes assigned to each sample. All of the reads were assigned to the corresponding samples by allowing one mismatch to the sample barcode and two mismatches to the adjacent PCR primer. The barcodes and primers were then trimmed, and the low quality sequences were removed. Reads longer than 200 bp were then aligned using a NAST-based[18]sequence aligner to a customized reference based on SILVA sequence alignment. We removed reads without any assignment in the anticipated region of the reference. Then, fi ltered reads were classif i ed using a Bayesian classif i er[19]with the RDP database (version 9, http://rdp.cme.msu. edu/). In order to obtain operational taxonomic units (OTUs), pairwise distances between high quality sequences were calculated and then clustered using Mothur, based on 97% similarity. The abundance and taxonomic classif i cation of each OTU in the sample were evaluated using Mothur. QIIME (Version 1.70, http://qiime.sourceforge.net/)[20]was used to analyze the distribution of all of the annotated OTUs (including the compositions and corresponding abundance) at different taxonomic levels (phylum, class, order, family, genus) for all of the samples. Metastats analysis was conducted to investigate the difference of the relative abundance for each species between the healthy and NAFLD groups.[21]APvalue less than 0.01 was used to def i ne signif i cant species differences.
Alpha-diversity was evaluated through fi ve indices (observed species, Chao, Simpson, Shannon, and ACE), as calculated with QIIME (version 1.70) based on the prof i le of the relative abundance of OTUs in each sample. All of the boxplot fi gures in this study were drawn by R (version 2.15.3). Principal Component Analysis (PCA) was performed using the ade4 package in R (version 2.15.3), based on the abundance of OTUs in each sample, to determine whether the healthy and NAFLD groups could be distinguished.
Results
Sample characteristics
Thirty-one patients underwent liver biopsies, and 26 controls were recruited within the study period. Three patients were excluded because of an inadequate liver biopsy sample size (<16 mm and/or <6 portal tracts). Two patients with chronic hepatitis B and one who had been taking antibiotics for three months were also excluded. In the control group, two had ALT and/or AST elevation, and two had a high LSM and/or CAP value. Thus, a total of 25 patients and 22 controls were ultimately analyzed. In addition, fi ve (20%) of the NAFLD patients had type 2 diabetes mellitus and six (24%) had hypertension. In the control group, only one subject had elevated fasting glucose levels. The characteristics of the study population are outlined in Table.
Data output and species complexity of the NAFLD patients
A total of 342 644 tags were produced for 47 samplesand subjected to standard fi ltration. After fi ltration, the average number of tags for all of the samples was 3880, and these tags were clustered into OTUs, which were used to determine microbial composition and corresponding abundance. Rarefaction analysis indicated that most of the diversity had been captured, although additional sequencing would have identif i ed some new phylotypes. The proportion of the classif i ed tags was more than 70% at the genus level, indicating the feasibility of conducting a comparative analysis on two groups at the genus level or higher. Alpha-diversity analysis showed a lower GM complexity in NAFLD compared to healthy subjects. We also found that healthy and NAFLD subjects could be signif i cantly distinguished as two different groups (Fig. 1).
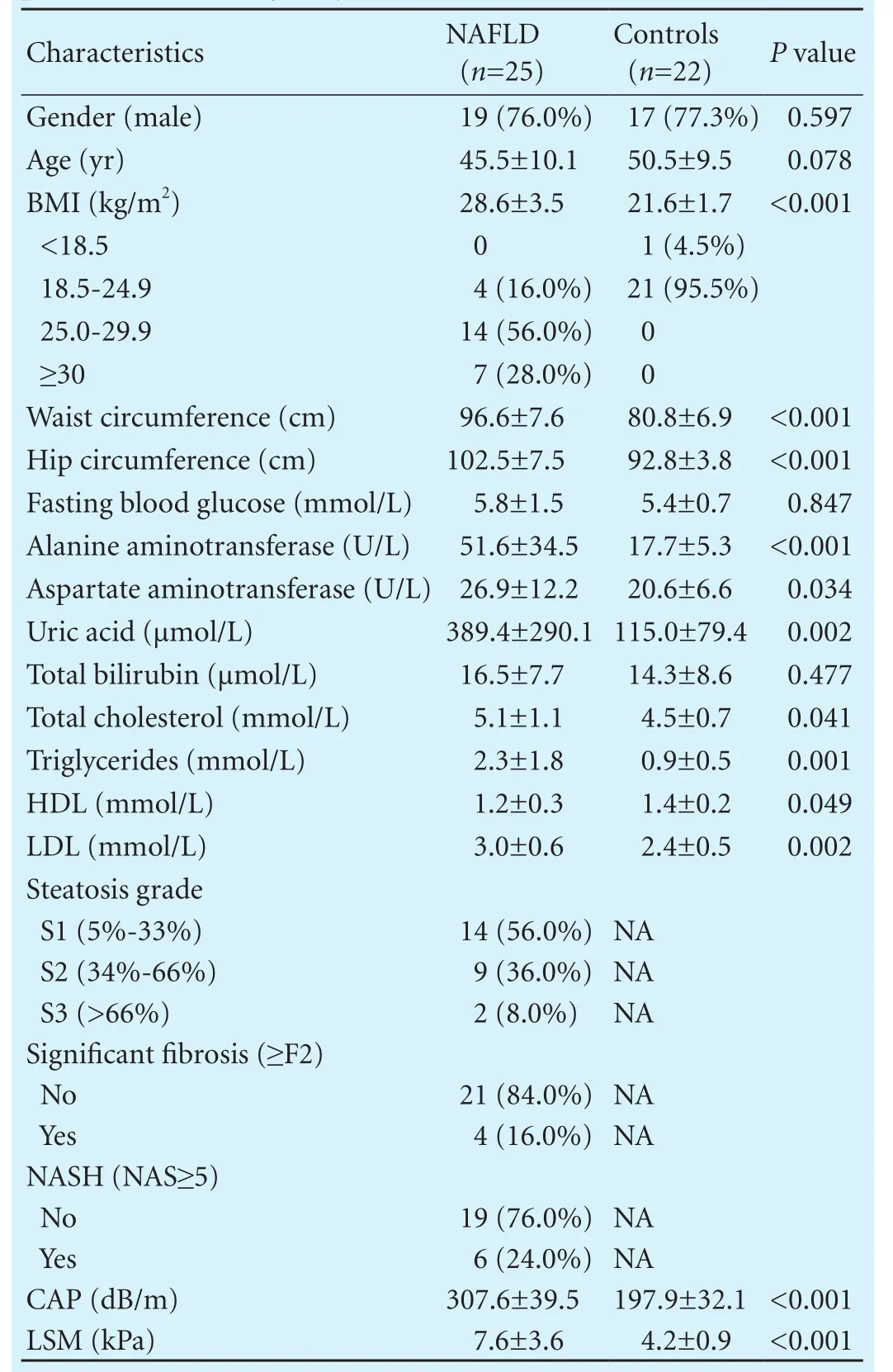
Table. Baseline and pathological characteristics of the NAFLD patients and healthy subjects
Enrichment of distinct microbes in the controls and NAFLD group
The NAFLD group was characterized by an apparent enrichment of Proteobacteria at 13.50% (P<0.01) abundance and Fusobacteria at 2.76% abundance (Fig. 2,P<0.01). The control group featured a high abundance of Bacteroidetes (44.63% compared to 26.55% in the NAFLD group) (P<0.01). The family Prevotellaceae accounted for a 28.66% abundance (P<0.01) in the control group, which was mainly attributed to the enrichment of genusPrevotella(27.56%,P<0.01). In contrast, the NAFLD group harbored a mere 6.23% and 5.83% abundance for the Prevotellaceae family andPrevotellagenus, respectively (P<0.01) (Figs. 3 and 4). The family Ruminococcaceae was also enriched in the control group (P<0.01) (26.44% vs 13.13% in NAFLD group), but only one known genus (Faecalibacterium) in this family accounts for >0.01%. Unclassif i ed genera also contributed to the distinctive species distribution between the NAFLD (11.69%) and control groups (21.63%,P<0.01).
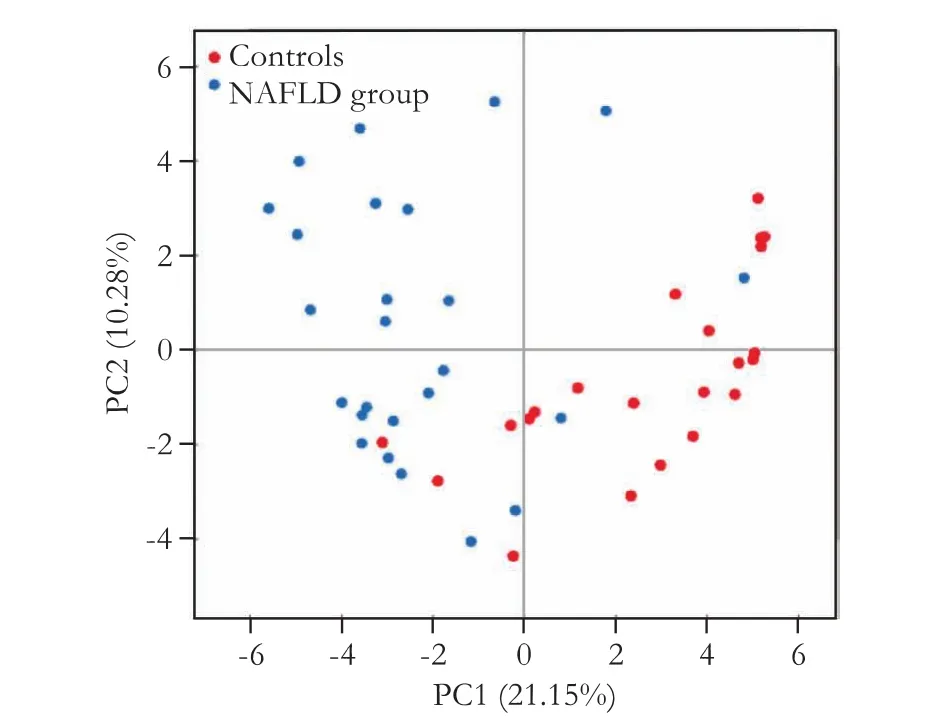
Fig. 1. Principal component analysis of the controls and NAFLD group. The healthy controls and the NAFLD group are clustered by signif i cantly different factors.
The family Lachnospiraceae had a 21.90% abundance in the NAFLD group as compared to an 11.86% abundance in the control group (P<0.01). The generaLachnospiraceae_Incertae_SedisandBlautia, classif i ed in the Lachnospiraceae family, showed a similar trend of more than two times the abundance in NAFLD patients versus healthy subjects. Another enriched species in the NAFLD group was the family Enterobacteriaceae, with a 12.02% (P<0.01) abundance, which was mainly attributed to genusEscherichia_Shigella(10.84%,P<0.01). The generaClostridium_XVIIIandStreptococcus, and the corresponding families Erysipelotrichaceae and Streptococcaceae, were all enriched in the NAFLD group (Figs. 3 and 4).
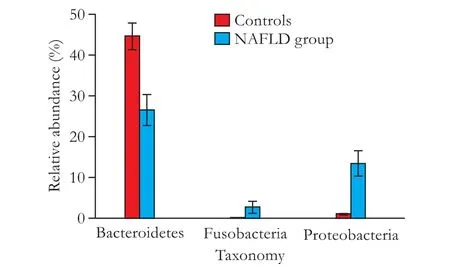
Fig. 2. Comparison of the mean relative abundances of phylotypes between the healthy controls and NAFLD group at the phylum level. A signif i cance threshold (P<0.01 and FDR<0.01) was chosen to conduct this analysis. Error bars indicate the standard deviations of the mean. The red box represents healthy controls and the blue box represents the NAFLD group.

Fig. 3. Comparison of the mean relative abundances of the phylotypes between the healthy controls and NAFLD group at the family level. A signif i cance threshold (P<0.01 and FDR<0.01) was chosen to conduct this analysis. Error bars indicate the standard deviations of the mean. The red box represents healthy controls and the blue box represents the NAFLD group.
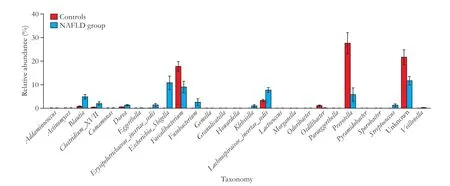
Fig. 4. Comparison of the mean relative abundances of the phylotypes between the healthy controls and NAFLD group at the genus level. A signif i cance threshold (P<0.01 and FDR<0.01) was chosen to perform analysis. Error bars indicate the standard deviations of the mean. The red box represents healthy controls and the blue box represents the NAFLD group.
Comparative analysis in patients with different BMIs
Since there were no overweight or obese subjects in the control group (Table), we selected the NAFLD group to identify discrepancies in the GM of subjects with different BMIs. The three subgroups (normal, overweight, and obese) could not be distinguished clearly based on GM composition and relative abundance, suggesting that there was no signif i cant difference among the three subgroups in NAFLD patients.
GM in patients with NASH or signif i cant fi brosis
A total of six (24%) patients were diagnosed with NASH according to NAS ≥5. Compared to those without NASH, patients with NASH had higher abundance of genusBlautia(5.82% vs 2.25%;P<0.01) and the corresponding Lachnospiraceae family (24.33% vs 14.21%;P<0.01).
Four patients (16%) had signif i cant fi brosis (F≥2) on liver biopsy. Compared to those with F0/F1 fi brosis, patients with signif i cant fi brosis had higher abundance of genusEscherichia_Shigella(12.53% vs 1.97%;P<0.01) and the corresponding Enterobacteriaceae family (13.92% vs 2.07%;P<0.01).
Discussion
The fi ndings of our study were similar to those of previous studies,[5,22,23]though we also found novel results at the genus level that await further analysis. We determined that NAFLD patients harbor lower GM diversity overall. At the phylum level, there is a predominance of Proteobacteria and Fusobacteria, from which most pathogens originate, in the GM of NAFLD patients. Healthy subjects harbor more Bacteroidetes in the gut, while NAFLD patients are enriched with the generaStreptococcus,Escherichia_Shigella,Lachnospiraceae_Ince rtae_Sedis, andBlautia. In addition, patients with NASH or signif i cant fi brosis (F≥2) possess a greater abundance of either genusBlautia(Lachnospiraceae family) or genusEscherichia_Shigella(Enterobacteriaceae family). The generaPrevotellaandFaecalibacteriumappear at decreased levels in NAFLD patients, which leads to a reduced accumulation of gut protectors and reduced intestinal energy harvesting capacity.
Healthy gut levels ofPrevotellaare well known to produce short chain fatty acids (SCFAs),[24]the typical agents of gut protection and energy assimilation. On the one hand, SCFAs and their G-protein coupled receptor 43 (GPR43) suppress colon inf l ammation and downregulate insulin signal transduction in adipose tissue.[25]Therefore, decreasedPrevotellaenrichment in NAFLD patients could lead to increased vulnerability of the gut surface and a weakening of the barrier function of the gut. On the other hand, SCFAs account for approximately 30% of dietary energy extraction and may contribute to the development of obesity. Therefore, the exact pathogenesis and impact of SCFAs on liver health remains unclear. Interestingly, a higher abundance ofPrevotellais a risk factor in children with obesity or NASH,[26]but may be a protective factor in adult NAFLD patients.[22]Indeed, a signif i cantly lower abundance ofPrevotellahas been found in adult cases of NASH and ≥F2 fi brosis.[27]These previous studies along with our results suggest that age may be the essential differentiating factor.
Faecalibacteriumalso tends to be enriched in healthy subjects, theFaecalibacteriumprausnitzii, a bacterium with anti-inf l ammatory properties, in particular wasshown to have increased from an undetectable percentage to a 14.5% abundance as health improved in humans.[28]On the other hand, sharply increasedEscherichia_Shigellain NAFLD patients can exacerbate gut leakiness by penetrating the intestinal epithelial barrier, which is a component of NAFLD pathogenesis.[5,23]Escherichia_Shigellaalso produce ethanol,[29]which enters the liver through blood circulation and causes disordered fatty acid metabolism.[30]The results of our study also ref l ected this positive association betweenEscherichia_Shigellaand NAFLD progression. However, the enrichment ofLachnospiraceae_Incertae_SedisandBlautiain the NASH group in our study was not consistent with previous reports. For example, several reports indicate that the family Lachnospiraceae contribute to the development of obesity and diabetes in ob/ob mice.[31,32]Other research found that the family Lachnospiraceae and genusBlautiawere enriched in healthy subjects compared to obese and NASH subjects.[7]Zhang et al[33]reported thatBlautiareduces inf l ammation levels and increases intestinal peristalsis. The discrepancies with respect to our fi ndings are most likely due to the age of the subjects; the previous studies involved children and young adults, whereas our study involved middleaged adults. Furthermore, the following may explain the increased levels ofBlautiain NAFLD patients: subtle homeostasis during NAFLD progression and the neutralization of impairment caused byEscherichia_Shigellaand other pathogens.
However, specif i c analysis to distinguish the normal, overweight, and obese subgroups in the NAFLD group did not yield positive results. This might be a type II error due to small case number in these subgroups or to the small contribution of obesity to NAFLD pathogenesis.[34]However, a greater number of NAFLD patients with varying BMI levels should be collected to analyze the contribution of BMI to GM in a future study.
In summary, we found that NAFLD patients tended to harbor lower GM diversity, which is known to impair GM homeostasis and disrupt the balance of energy harvesting and microbial metabolites. The pathogens are derived mainly from the Proteobacteria and Fusobacteria phyla, leading to increased levels of microbial gut toxins in NAFLD patients. We infer that this may be the reason why the abundance ofLachnospiraceae_Incertae_SedisandBlautiais elevated in the NAFLD or NASH groups, since these two genera were shown to regulate intestinal inf l ammation and peristalsis. DecreasedPrevotellamight lead to fewer SCFAs in the NAFLD patient’s gut, thereby making the gut mucosa more susceptible to impairment by the metabolites of pathogens in adults. IncreasedEscherichia_Shigellamight also exacerbate gut leakiness due to the robust production of ethanol. As a result, more toxins and alcohol enter into the blood and lymph cycle, disturbing lipid metabolism in the liver.
The fi ndings in this study, as well as other studies, are critical to the understanding of the complex contribution of GM to NAFLD pathogenesis. Most importantly, these studies will help uncover the role of specif i c bacteria, such asLachnospiraceae_Incertae_Sedis, and other unclassif i ed genera in NAFLD pathogenesis.[22,31]Further research is necessary to clarify the relevance of the gut microbial taxa discovered in our work to the pathogenesis and progression of human NAFLD.
Contributors: SF, ZRD and FJG proposed the study. SF, DWJ and WXY performed the research. ZRD and SXQ carried out the statistical analysis. SF and FJG wrote the fi rst draft. All authors contributed to the design and interpretation of the study and to further drafts. FJG is the guarantor.
Funding: This study was supported by grants from the National Key Basic Research Project (2012CB517501), the Chinese Foundation for Hepatitis Prevention and Control -- “Wang Bao-En” Liver Fibrosis Research Foundation (XJS20120501), and the National Natural Science Foundation of China (81400610).
Ethical approval: This study was approved by the Ethics Committees of Xinhua Hospital (XHEC-C-2012-023).
Competing interest: No benef i ts in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686-690.
2 Aron-Wisnewsky J, Gaborit B, Dutour A, Clement K. Gut microbiota and non-alcoholic fatty liver disease: new insights. Clin Microbiol Infect 2013;19:338-348.
3 Gangarapu V, Yıldız K, Ince AT, Baysal B. Role of gut microbiota: obesity and NAFLD. Turk J Gastroenterol 2014;25:133-140.
4 Compare D, Coccoli P, Rocco A, Nardone OM, De Maria S, Cartenì M, et al. Gut--liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis 2012;22:471-476.
5 Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009;49:1877-1887.
6 Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, et al. Elevated endotoxin levels in nonalcoholic fatty liver disease. J Inf l amm (Lond) 2010;7:15.
7 Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 2013;57:601-609.
8 Go GW, Oh S, Park M, Gang G, McLean D, Yang HS, et al. t10,c12 conjugated linoleic acid upregulates hepaticde novolipogenesis and triglyceride synthesis via mTOR pathway activation. J Microbiol Biotechnol 2013;23:1569-1576.
9 Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, et al. Metabolic prof i ling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A 2006;103:12511-12516.
10 Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010;328:228-231.
11 De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, et al. Dysbiosis contributes to fi brogenesis in the course of chronic liver injury in mice. Hepatology 2014;59:1738-1749.
12 Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013;500:541-546.
13 Fan JG, Jia JD, Li YM, Wang BY, Lu LG, Shi JP, et al. Guidelines for the diagnosis and management of nonalcoholic fatty liver disease: update 2010: (published in Chinese on Chinese Journal of Hepatology 2010; 18:163-166). J Dig Dis 2011;12:38-44.
14 Wong VW, Vergniol J, Wong GL, Foucher J, Chan HL, Le Bail B, et al. Diagnosis of fi brosis and cirrhosis using liver stiffness measurement in nonalcoholic fatty liver disease. Hepatology 2010;51:454-462.
15 Karlas T, Petroff D, Garnov N, Böhm S, Tenckhoff H, Wittekind C, et al. Non-invasive assessment of hepatic steatosis in patients with NAFLD using controlled attenuation parameter and 1H-MR spectroscopy. PLoS One 2014;9:e91987.
16 Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313-1321.
17 Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platformindependent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009;75:7537-7541.
18 DeSantis TZ Jr, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, et al. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 2006;34:W394-399.
19 Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifi er for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007;73:5261-5267.
20 Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7:335-336.
21 White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol 2009;5:e1000352.
22 Jiang W, Wu N, Wang X, Chi Y, Zhang Y, Qiu X, et al. Dysbiosis gut microbiota associated with inf l ammation and impaired mucosal immune function in intestine of humans with nonalcoholic fatty liver disease. Sci Rep 2015;5:8096.
23 Croxen MA, Law RJ, Scholz R, Keeney KM, Wlodarska M, Finlay BB. Recent advances in understanding enteric pathogenic Escherichia coli. Clin Microbiol Rev 2013;26:822-880.
24 Purushe J, Fouts DE, Morrison M, White BA, Mackie RI; North American Consortium for Rumen Bacteria, et al. Comparative genome analysis ofPrevotellaruminicola andPrevotellabryantii: insights into their environmental niche. Microb Ecol 2010;60:721-729.
25 Zhu L, Baker RD, Baker SS. Gut microbiome and nonalcoholic fatty liver diseases. Pediatr Res 2015;77:245-251.
26 Abdou RM, Zhu L, Baker RD, Baker SS. Gut microbiota of nonalcoholic fatty liver disease. Dig Dis Sci 2016;61:1268-1281.
27 Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016;63:764-775.
28 Hvistendahl M. My microbiome and me. Science 2012;336:1248-1250.
29 Clark DP. The fermentation pathways of Escherichia coli. FEMS Microbiol Rev 1989;5:223-234.
30 Liu J. Ethanol and liver: recent insights into the mechanisms of ethanol-induced fatty liver. World J Gastroenterol 2014;20:14672-14685.
31 Kameyama K, Itoh K. Intestinal colonization by a Lachnospiraceae bacterium contributes to the development of diabetes in obese mice. Microbes Environ 2014;29:427-430.
32 Zeng H, Ishaq SL, Zhao FQ, Wright AD. Colonic inf l ammation accompanies an increase of β-catenin signaling and Lachnospiraceae/Streptococcaceae bacteria in the hind gut of high-fat diet-fed mice. J Nutr Biochem 2016;35:30-36.
33 Zhang J, Guo Z, Xue Z, Sun Z, Zhang M, Wang L, et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. ISME J 2015;9:1979-1990.
34 Relman DA. Restoration of the gut microbial habitat as a disease therapy. Nat Biotechnol 2013;31:35-37.
May 2, 2016
Accepted after revision November 30, 2016
Author Aff i liations: Department of Gastroenterology (Shen F, Ding WJ and Fan JG), Department of Pathology (Wang XY), Xinhua Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200092, China; Research and Therapy Centre for Liver Disease, Zhengxing Hospital, Zhangzhou 363000, China (Zheng RD); Department of Microbial Genomics Research, BGI Shenzhen, Shenzhen 518083, China (Sun XQ)
Jian-Gao Fan, MD, PhD, Department of Gastroenterology, Xinhua Hospital, Shanghai Jiaotong University School of Medicine, 1665 Kongjiang Road, Shanghai 200092, China (Tel/Fax:+86-21-25077340; Email: Fanjiangao@xinhuamed.com.cn)
© 2017, Hepatobiliary Pancreat Dis Int. All rights reserved.
10.1016/S1499-3872(17)60019-5
Published online May 26, 2017.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Predictive value of C-reactive protein/albumin ratio in acute pancreatitis
- The International Study Group of Pancreatic Surgery def i nition of delayed gastric emptying and the effects of various surgical modif i cations on the occurrence of delayed gastric emptying after pancreatoduodenectomy
- Hepatopancreatoduodenectomy for advanced hepatobiliary malignancies: a single-center experience
- Interaction between insulin-like growth factor binding protein-related protein 1 and transforming growth factor beta 1 in primary hepatic stellate cells
- Bilioenteric anastomotic stricture in patients with benign and malignant tumors: prevalence, risk factors and treatment
- Effects of multimodal fast-track surgery on liver transplantation outcomes