ZnFe2O4/聚苯胺复合材料的制备及光催化性能*
2017-08-02冒卫星赵巧云高晓宇沈欣怡童国秀吕天喜宫培军
冉 方, 冒卫星, 赵巧云, 高晓宇, 沈欣怡,童国秀, 吕天喜, 宫培军
(浙江师范大学 化学与生命科学学院,浙江 金华 321004)
ZnFe2O4/聚苯胺复合材料的制备及光催化性能*
冉 方, 冒卫星, 赵巧云, 高晓宇, 沈欣怡,童国秀, 吕天喜, 宫培军
(浙江师范大学 化学与生命科学学院,浙江 金华 321004)
采用化学氧化聚合法制备了组分含量比不同的ZnFe2O4/聚苯胺纳米复合材料(ZnFe2O4/PANI),对产物进行了表征,并研究了其对罗丹明B的吸附性能和可见光下的光催化性能.结果表明:随着纳米ZnFe2O4与苯胺的投料质量比(wZ/A)的减小,ZnFe2O4/PANI中聚苯胺(PANI)的质量分数和酸掺杂度均增大,复合物的可见光吸收性能和导电性均增强;同时,PANI的修饰提高了催化剂对罗丹明B的吸附能力,平衡吸附量随着wZ/A的减小而缓慢增加;产物的光催化性能先增加、后减小,wZ/A=10%的产物的光催化性能最好,且在重复使用5次内呈现良好的催化稳定性,可磁回收.结合活性物种捕获实验结果,推测其光催化反应过程中氧化性自由基和空穴共同参与了罗丹明B的降解.
铁酸锌;聚苯胺;化学氧化聚合法;光催化;磁响应性
近二十多年来,半导体光催化技术在有机污染物废水降解方面的应用研究受到国内外学者的关注.研发经济性好、稳定性佳、可见光催化活性高的半导体光催化剂是实现光催化技术走向实际应用的前提.
铁酸锌(ZnFe2O4)是一种窄带隙(约1.9 eV)、对可见光响应的n型半导体材料,具有无毒、化学稳定性好、耐光化学腐蚀、制备简便和成本低等特点[1],其磁响应性为催化剂的回收提供了方便[2].然而,ZnFe2O4的不足在于光生载流子的迁移距离短、光生电子和空穴容易复合,因而光催化活性较低.因此,利用ZnFe2O4与能带位置匹配的其他半导体复合构建半导体复合材料,成为开发ZnFe2O4基光催化剂的重要策略[3].
聚苯胺(PANI)是一种化学稳定性好、电导率可调、可见光吸收性能强、成本低的导电聚合物,能作为电子供体和空穴导体促进光催化剂的光生载流子的有效分离,同时可改善TiO2等宽带隙光催化剂对可见光的吸收[4].文献[5]采用吸附法制备了TiO2/PANI复合光催化剂,发现当PANI/TiO2质量比为3.0%时产物的光催化活性最佳.该方法制备的催化剂中PANI的质量分数普遍很低(不足5%).敏世雄等[6]在硅烷偶联剂改性的纳米TiO2表面接枝聚合苯胺单体,获得了PANI含量可调幅度较大的TiO2/PANI复合材料,但催化剂在重复使用中活性降低较明显.文献[7]采用化学氧化聚合法制备了CoFe2O4/PANI磁性复合光催化剂,催化剂对阴离子和中性有机染料的吸附加速了染料的光催化降解进程.
基于PANI的最高占据轨道(HOMO)和最低空轨道(LUMO)电位与ZnFe2O4的价带(VB)和导带(CB)电位匹配,通过PANI对ZnFe2O4的修饰有望提高后者的光生载流子的分离效率,从而可获得光催化活性较好的光催化剂.关于ZnFe2O4/PANI复合材料的制备与微波吸收/屏蔽性能的研究已有很多报道[8-9],但关于ZnFe2O4/PANI的组成影响其吸附和光催化性能的研究却罕有报道.因此,本文采用化学氧化聚合法制备了一系列的ZnFe2O4/PANI复合光催化剂,研究了该催化剂的组成与其光学吸收性质、染料吸附和光催化性能之间的关系,并推测了其光催化反应的可能机理.
1 实验部分
1.1 试剂
所有试剂均购自国药集团化学试剂有限公司,分析纯.苯胺(ANI)在使用前经减压蒸馏纯化,其他试剂直接使用.
1.2 纳米ZnFe2O4的制备
取1 mmol Zn(NO3)2·6H2O和2 mmol Fe(NO3)3·9H2O溶解在100 mL蒸馏水中,快速搅拌下滴加6 mol·L-1NaOH溶液至pH值为12,在80 ℃下反应2 h.将固体产物过滤并水洗数次,干燥后在600 ℃下煅烧2 h,得到纳米ZnFe2O4.
1.3 ZnFe2O4/PANI复合光催化剂的制备
将500 mg纳米ZnFe2O4分散在100 mL HCl溶液(0.1 mol·L-1)中,加入50 mg苯胺,混匀后缓慢滴入10 mL 过硫酸铵的 HCl溶液(0.1 mol·L-1),在0 ℃下搅拌反应6 h,抽滤,用水洗涤固体产物至滤液无色,60 ℃真空干燥24 h,得到ZnFe2O4与苯胺投料质量比(wZ/A)为10的ZnFe2O4/PANI复合催化剂,标记为Z/P-10.固定ZnFe2O4的用量,调整苯胺的用量,采用相同方法制备wZ/A分别为20,5和2.5的ZnFe2O4/PANI,依次标记为Z/P-20,Z/P-5和Z/P-2.5.
1.4 产物的表征
XRD采用PW3040/60型X-射线衍射仪(荷兰Philps公司)测定;FTIR谱使用Nexus 670型傅里叶变换红外光谱仪(美国Nicolet公司)测定;TGA在STA 449 C型热分析仪(德国Netzsch公司)上获得,空气气氛,升温速率为10 ℃·min-1;TEM借助JEOL-2010型透射电子显微镜(日本JEOE公司)观测;UV-Vis DRS由Evolution 500LC型紫外-可见分光光度计(美国Thermo 公司)测定.催化剂的表面化学环境借助ESCALAB 250Xi型X射线光电子能谱仪(XPS,美国Thermo Fisher Scientific公司)测定,磁学性能用7404型振动样品磁强计(美国Lakeshore 公司)测定,测试温度为25 ℃.
1.5 催化剂的吸附性能
在50 mL石英烧杯中加入30 mL罗丹明B(RhB)溶液(5 mg·L-1)和50 mg催化剂样品,在磁力搅拌下黑暗吸附,将不同时刻取出的3 mL反应液离心以去除催化剂,借助UV-2700型紫外-可见分光光度计(日本Shimadzu公司)测定上清液在554 nm波长处的吸光度(A),借助RhB溶液的吸光度-浓度标准曲线求出染料的浓度Ct,计算出该时刻单位质量催化剂对RhB的吸附量,以吸附量-时间作图获得吸附动力学曲线,将吸附量值达到稳定的时刻作为平衡吸附时间.
1.6 催化剂的光催化性能
自制光催化光源[9]装配3只8 W日光灯管,经ORION-TH型激光功率能量计测定反应液面处的辐照度为0.50 mW·cm-2.将含有50 mg催化剂的30 mL RhB溶液(5 mg·L-1)在黑暗下搅拌120 min后置于日光灯下辐照,开始辐照时刻为0 min,每30 min取样3 mL测定上清液在554 nm波长处的吸光度,得出RhB的浓度Ct,以Ct与0 min时RhB的浓度C0的比值(Ct/C0)与1的差值表示RhB的降解率.在捕获实验中,光照反应前分别加入叔丁醇(t-BuOH)或乙二胺四乙酸二钠(EDTA-2Na),其他条件不变,测试反应体系中RhB的降解率.
2 结果与讨论
2.1 产物的结构和化学组成分析
图1(a)为ZnFe2O4,PANI和ZnFe2O4/PANI的XRD谱图.ZnFe2O4样品的衍射峰与立方尖晶石结构ZnFe2O4的标准图卡(JCPDS 89-1012)吻合.PANI在20.6°和25.5°附近有2个宽衍射峰,对应于高分子链段周期性平行和垂直排列的衍射信号[4].ZnFe2O4/PANI的XRD谱中ZnFe2O4的衍射峰均清晰可见,但在20.6°和25.5°的衍射峰因强度很低而难以观测.这是因为ZnFe2O4与PANI相互作用导致PANI链段的有序排列受限,因而结晶度降低[10].
借助TGA确定了ZnFe2O4/PANI中PANI的质量分数(见图2).与PANI的热分解过程(见图2插图)的相似之处在于:ZnFe2O4/PANI在150 ℃前

(a)XRD (b)FTIR a:ZnFe2O4;b:PANI;c:Z/P-20;d:Z/P-10;e:Z/P-5;f:Z/P-2.5图1 样品的XRD谱图及FTIR谱图
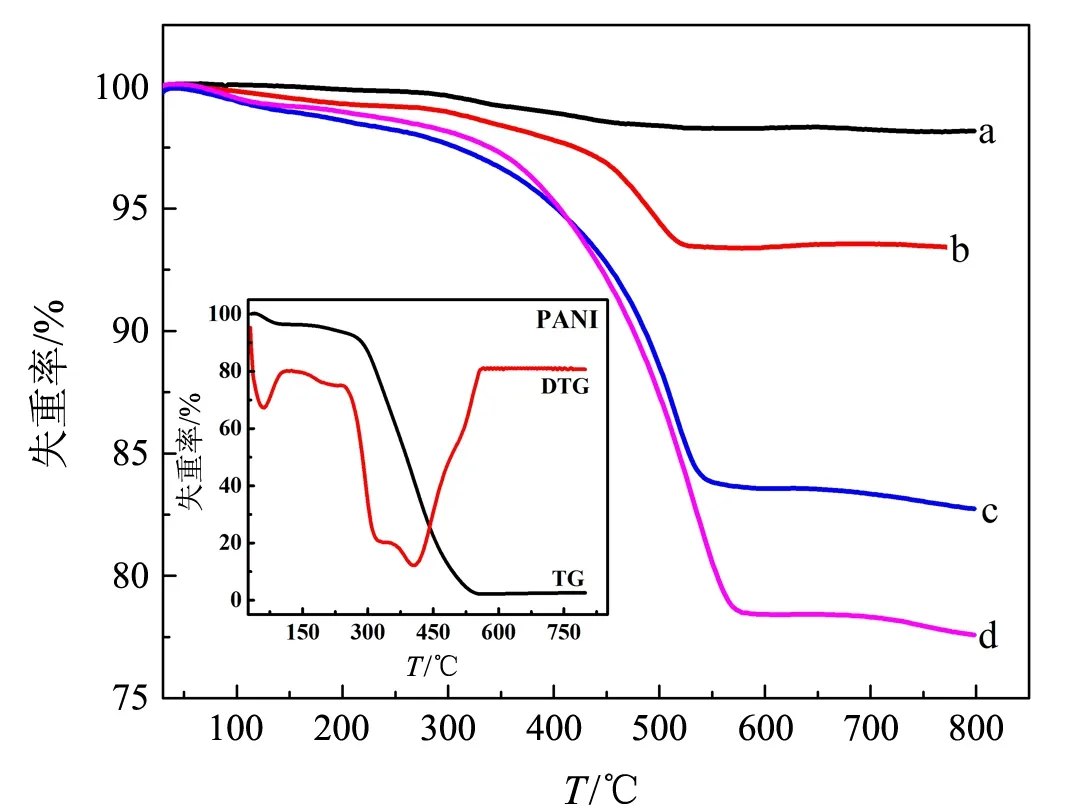
a:Z/P-20;b:Z/P-10;c:Z/P-5;d:Z/P-2.5图2 ZnFe2O4/PANI 和PANI(插图)的热重曲线
的失重源于样品中少量吸附水的蒸发,此后缓慢的失重对应于部分掺杂酸(HCl)的挥发,在240 ℃以上PANI链开始快速分解;差别在于:PANI的完全分解温度约为560 ℃,而复合物中PANI的完全分解温度依次约为590,600,610和620 ℃,呈现随wZ/A的减小逐渐升高的趋势.推测ZnFe2O4与PANI之间的相互作用限制了PANI分子链的热运动幅度[13],延缓了与ZnFe2O4表面直接作用的PANI链段的热分解进程.TGA分析确定Z/P-20,Z/P-10,Z/P-5和Z/P-2.5中PANI的质量分数依次为1.8%,6.6%,16.4%和21.6%.
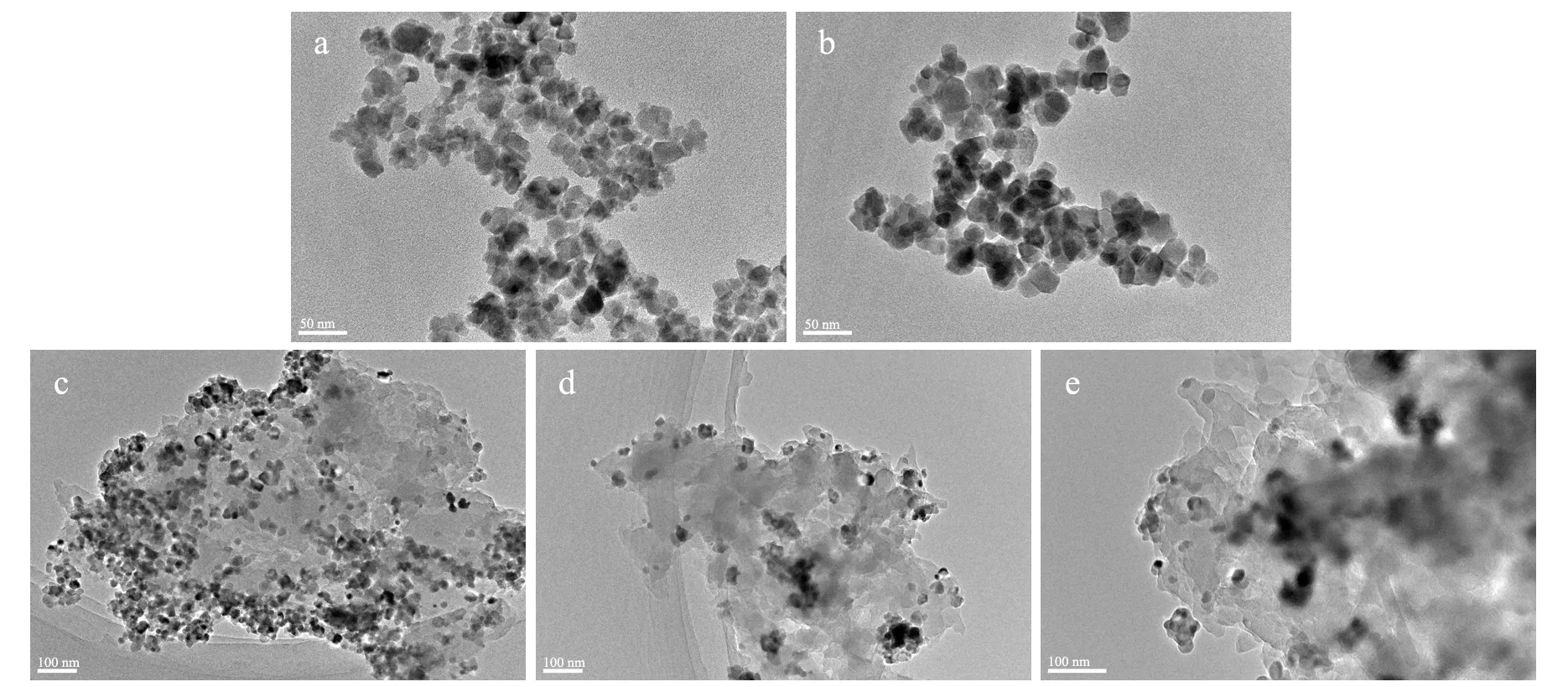
a:ZnFe2O4;b:Z/P-20;c:Z/P-10;d:Z/P-5;e:Z/P-2.5.图3 样品的TEM图
2.2 产物的形貌分析
TEM分析(见图3)显示,ZnFe2O4的尺寸为15~25 nm,PANI含量低的Z/P-20在形貌上与ZnFe2O4无明显的差别.当wZ/A进一步减小时,ZnFe2O4/PANI演变至微米级团状物,处于团状PANI表层且聚集的ZnFe2O4粒子逐渐趋向于均匀分散在PANI团内的深处.ZnFe2O4与PANI链的充分接触可以使组分间的相互作用增强,也有利于光生载流子在两组分间的传递.因为PANI能通过静电作用或π-π作用吸附含有异性电荷或共轭结构的有机分子[7],所以,PANI的适度修饰可以促进光催化剂对含上述结构的有机分子的富集,加速其去除进程;但是,过厚且致密的PANI包覆层会阻碍有机分子向ZnFe2O4的表面迁移,从而降低催化剂的光催化性能[6].因此,本实验中未制备PANI含量更高的ZnFe2O4/PANI.
2.3 产物的UV-Vis DRS分析
紫外-可见漫反射谱(见图4)显示:ZnFe2O4
吸收紫外光和680 nm波长以下的可见光,拟合可得其禁带宽度约为1.80 eV,比文献[14]报道的ZnFe2O4的禁带宽度(2.3 eV)略小;PANI在200~800 nm波长处均有吸收,其中400~800 nm宽而强的吸收带(λmax约为 615 nm)对应于主链的π-π*跃迁[6];ZnFe2O4/PANI对可见光的吸收
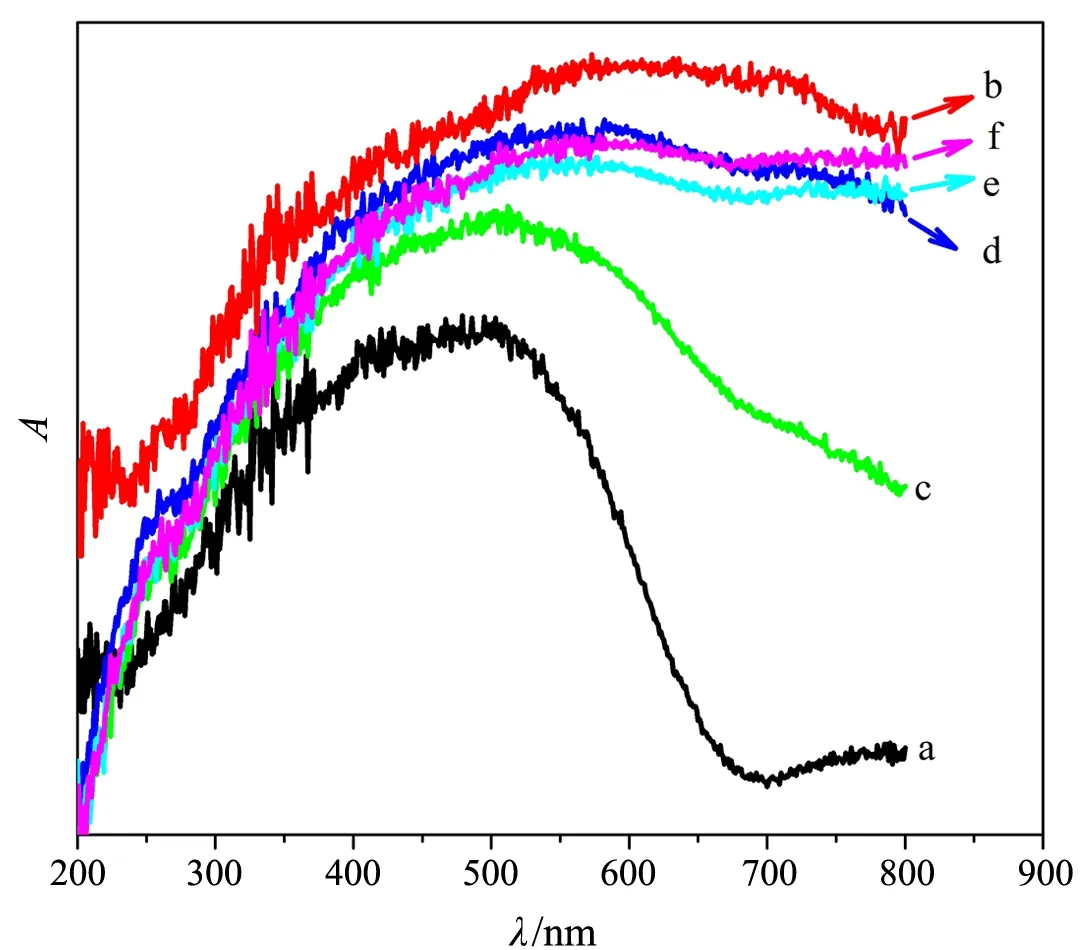
a:ZnFe2O4;b:PANI;c:Z/P-20;d:Z/P-10;e:Z/P-5;f:Z/P-2.5图4 样品的UV-Vis DRS图
强度随wZ/A的减小而逐渐增大,表明PANI的修饰改善了催化剂对长波可见光的吸收性能,这是ZnFe2O4/PANI高效利用太阳光催化降解有机染料的重要前提.PANI质量分数最低的Z/P-20的λmax最小,意味着该材料中PANI的共轭链长度较其他ZnFe2O4/PANI的短.
2.4 产物的电导率分析
测试了各样品的导电性能.极低的电导率(3.41×10-10S·cm-1)表明ZnFe2O4几乎不导电.
Z/P-20,Z/P-10,Z/P-5和Z/P-2.5的电导率依次为1.53×10-9,1.57×10-8,8.07×10-7和8.67×10-7S·cm-1,随着wZ/A的减小而逐渐增大,与复合物所含PANI的酸掺杂率和质量分数均逐渐增大的趋势一致.本实验制备的PANI的电导率为6.94×10-5S·cm-1,其值较文献[11,15]报道的具有高掺杂率的PANI的电导率(10-1数量级)低4~5个数量级,而本征态PANI则不导电.结合FTIR分析结果,推测ZnFe2O4/PANI中PANI的掺杂率总体上较低,这可能不利于快速传导和分离光生载流子.
2.5 催化剂的吸附及光催化性能
图5(a)给出了PANI,ZnFe2O4和ZnFe2O4/PANI对RhB的吸附平衡曲线.所有样品均在120 min内达到吸附平衡,ZnFe2O4和PANI对RhB的平衡吸附量分别为0.50和1.58 mg/g;复合物的平衡吸附量依次为1.67,1.73,1.85和2.14 mg/g,随wZ/A的减小而增大,但总体上较ZnFe2O4的增幅不大.结合XRD,FTIR和电导率的分析,笔者认为产生这一现象的原因在于PANI与阳离子型染料RhB的结合方式,包括:1)PANI中苯环、醌环等共轭结构对RhB分子中的苯环等富电子结构单元的吸引能力,它随样品中PANI
质量分数的增大而增强;2)PANI骨架中正电性的酸掺杂位点N+对RhB中正电荷中心N+的静电排斥力[7],它随样品中PANI的酸掺杂度的增大而增强.对于纯PANI,相对高的结晶度和酸掺杂程度使得链段排列较有序、正电性较强,不利于RhB在其表面的吸附.而在复合物中,纳米半导体的存在一定程度上阻碍了PANI链的规则排列,有利于RhB与PANI链段的充分接触,其所含PANI的酸掺杂度相对较低,因此更有利于RhB的吸附;随着wZ/A的减小,两种效果相反的作用相互叠加,最终使ZnFe2O4/PANI对RhB的吸附能力总体上相差不大,仅轻微提高.
图5(b)显示了催化剂在可见光下催化降解RhB的结果.无催化剂时,可见光对RhB的光解作用很弱,150 min内约3.0%的RhB发生分解.相比PANI和ZnFe2O4分别对RhB的光催化降解率12.3%和62.0%,ZnFe2O4/PANI存在下RhB的降解率依次为42.7%,85.4%,63.4%和43.7%,随wZ/A的减小呈现先增大、后减小的趋势,Z/P-10的光催化性能最优.结合上述分析,笔者认为导致ZnFe2O4/PANI光催化性能变化趋势的可能的原因是:当wZ/A较大时,ZnFe2O4表面的PANI包覆层以导电性很差的短共轭链为主,光生电子和空穴难以在PANI和ZnFe2O4之间迁移[6],不能提高光生载流子的分离效率;相比之下,PANI覆盖使染料分子与ZnFe2O4表面的直接接触变得困难,带给催化性能的负面作用较显著,导致Z/P-20的光催化性能低于纳米ZnFe2O4.当wZ/A适当减小时,复合材料中PANI的质量分数、酸掺杂度和共轭链长度均增大,PANI与ZnFe2O4之间传导光生载流子的能力得以增强,对光催化性能的促进作用大于ZnFe2O4表面被覆盖的负面
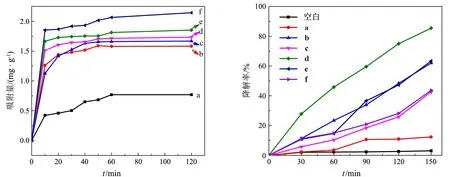
(a)吸附动力学曲线 (b)光催化降解曲线a:ZnFe2O4;b:PANI;c:Z/P-20;d:Z/P-10;e:Z/P-5;f:Z/P-2.5图5 催化剂对RhB的吸附动力学曲线及光催化降解曲线
作用,Z/P-10的光催化活性得以优化,其活性优于文献[7]报道的CoFe2O4/PANI复合光催化剂(可见光光照9 h,RhB降解率约为30%).当wZ/A进一步减小时,RhB迁移到深埋于PANI微米团内部的ZnFe2O4表面的难度增大[6];同时,PANI和被吸附的RhB很强的光子吸收能力导致半导体粒子难以接受光子,无法发挥光催化作用[16],因此Z/P-5和Z/P-2.5的光催化性能降低.
2.6 催化剂的重复使用性能
良好的催化稳定性是光催化剂实际应用于有机废水降解的重要前提.图6(a)显示,当Z/P-10催化剂循环使用5次时,RhB的降解率仍高于70%,表明催化剂的催化稳定性良好.对于导致催化剂活性轻微下降的原因,X射线光电子能谱分析(图略)显示,催化反应后催化剂的O元素结合能谱在531.03 eV处的吸收峰面积减小,表明ZnFe2O4表面的—OH等物种[17]在光催化过程中被部分消耗,推测ZnFe2O4表面—OH数目的减少可能是ZnFe2O4/PANI催化活性下降的主要原因.此外,尽管回收催化剂的FTIR谱中未出现RhB或其结构单元对应的特征吸收带,但不排除存在微量的RhB降解产物吸附在催化剂表面,导致催化剂的活性下降[18].
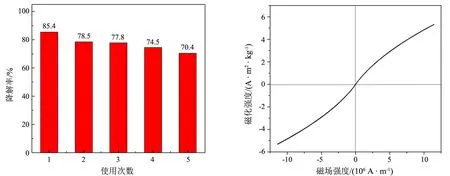
(a)重复使用曲线 (b)磁滞回线 图6 Zn/P-10的重复使用性能和磁滞回线
图6(b)为Z/P-10的室温磁滞回线,磁化曲线没有磁滞现象,矫顽力和剩余磁化强度均接近于0,表现为顺磁性.样品的磁化强度(M)在测试磁场强度范围内未达到饱和,当磁场强度为1.14×107A·m-1时,M为5.32 A·m2·g-1,这一数值介于文献[2-3]报道的ZnFe2O4磁性复合光催化剂的M值之间.实验显示,借助磁铁可以在10 min内从RhB溶液中回收催化剂,所得催化剂可以经搅拌再次分散到RhB溶液中.良好的磁响应性有助于Z/P-10的回收和高效再利用.
2.7 催化剂的光催化机理
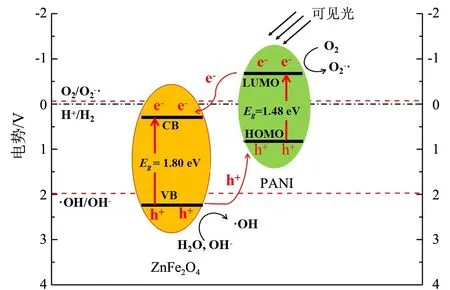
图7 ZnFe2O4/PANI光催化剂的载流子迁移示意图
3 结 论
采用化学氧化聚合法制备了组分含量比不同的ZnFe2O4/PANI复合光催化剂.随着催化剂中PANI质量分数的增大,PANI的酸掺杂度逐渐提高,复合物的导电性增强,ZnFe2O4更均匀地散布在PANI内部,这些特征均有利于光生载流子在PANI与ZnFe2O4之间的有效迁移.同时,PANI的复合拓宽了ZnFe2O4对可见光的吸收范围,在一定程度上提高了催化剂对RhB的吸附能力.ZnFe2O4/PANI的光催化性能呈现先增大、后减小的趋势,其中Z/P-10的光催化性能最好,重复使用性能和磁响应性良好.ZnFe2O4表面—OH的消耗可能是催化活性轻微降低的主要原因.捕获实验证实,在可见光催化过程中氧化性自由基和空穴共同参与了RhB的降解.
[1]Tong Guoxiu,Du Fangfang,Wu Wenhua,et al.Enhanced reactive oxygen species (ROS) yields and antibacterial activity of spongy ZnO/ZnFe2O4hybrid micro-hexahedra selectively synthesized through a versatile glucose-engineered co-precipitation/annealing process[J].J Mater Chem B,2013,1(20):2647-2657.
[2]Zhang Shouwei,Li Jiaxing,Zeng Meiyi,et al.In situ synthesis of water-soluble magnetic graphitic carbon nitride photocatalyst and its synergistic catalytic performance[J].ACS Appl Mater Interfaces,2013,5(23):12735-12743.
[3]Zhang Wanqing,Wang Mei,Zhao Wenji.Magnetic composite photocatalyst ZnFe2O4/BiVO4:Synthesis,characterization,and visible-light photocatalytic activity[J].Dalton Trans,2013,42:15464-15474.
[4]Guo Na,Liang Yimai,Lan Shi,et al.Microscale hierarchical three-dimensional flowerlike TiO2/PANI composite:Synthesis,characterization,and its remarkable photocatalytic activity on organic dyes under UV-light and sunlight irradiation[J].J Phys Chem C,2014,118:18343-18355.
[5]Zhang Hao,Zong Ruilong,Zhao Jincai,et al.Dramatic visible photocatalytic degradation performances due to synergetic effect of TiO2with PANI[J].Environ Sci Technol,2008,42(10):3803-3807.
[6]敏世雄,王芳,张振敏,等.PANI/AMTES-TiO2纳米复合材料的制备及其光催化性能[J].物理化学学报,2009,25(7):1303-1310.
[7]Xiong Pan,Chen Qun,He Mingyang,et al.Cobalt ferrite-polyaniline heteroarchitecture:A magnetically recyclable photocatalyst with highly enhanced performances[J].J Mater Chem,2012,22(34):17485-17493.
[8]李涓碧,吴雪,李良超,等.Zn-Ni-Cu 铁氧体/聚邻甲氧基苯胺复合物的制备及电磁性能[J].中国科学 B辑:化学,2014,44(3):309-319.
[9]穆肖,宓雨泽,肖立伟,等.Fe3O4/聚苯胺纳米纤维的紫外光辅助化学氧化聚合法合成[J].浙江师范大学学报:自然科学版,2015,38(5):426-434.
[10]Li Xingwei,Wang Gengchao,Li Xiaoxuan,et al.Surface properties of polyaniline/nano-TiO2composites[J].Appl Surf Sci,2004,229(1):395-401.
[11]孙红娟,胡傲厚,彭同江.pH 值对聚苯胺的结构和导电性能的影响[J].西南科技大学学报,2011,26(1):28-32.
[12]Wang Ning,Li Jingjing,Lü Wei,et al.Synthesis of polyaniline/TiO2composite with excellent adsorption performance on acid red G[J].RSC Adv,2015,5:21132-21141.
[13]李宪华,张雷刚,王雪雪,等.界面聚合法制备PANI/g-C3N4复合催化剂及其热稳定性和可见光催化性能[J].物理化学学报,2015,31(4):764-770.
[14]Maryam M,Fahimeh K C,Nahid R,et al.ZnFe2O4nanoparticle:Synthesis and photocatalytic activity under UV-Vis and visible light[J].Iranian Chem Comm,2015,3:166-173.
[15]唐劲松,王利祥,景遐斌,等.酸浓度对苯胺聚合及所得产物的结构与性能的影响[J].高分子学报,1989(2):188-192.
[16]Bu Yuyu,Chen Zhuoyuan.Role of polyaniline on the photocatalytic degradation and stability performance of the polyaniline/silver/silver phosphate composite under visible light[J].ACS Appl Mater Interfaces,2014,6:17589-17598.
[17]Takashi K,Satoshi S,Takashi K,et al.Photoemission study of dissociatively adsorbed methane on a pre-oxidized SnO2thin film[J].Surf Sci,2000,448(2/3):101-107.
[18]Liu Zhenyan,Miao Yuee,Liu Mingkai,et al.Flexible polyaniline-coated TiO2/SiO2nanofiber membranes with enhanced visible-light photocatalytic degradation performance[J].J Colloid Interface Sci,2014,424:49-55.
[19]Kong Liang,Jiang Zheng,Xiao Tiancun,et al.Exceptional visible-light-driven photocatalytic activity over BiOBr-ZnFe2O4heterojunctions[J].Chem Commun,2011,47:5512-5514.
[20]Belabed C,Abdi A,Benabdelghani Z,et al.Photoelectrochemical properties of doped polyaniline:Application to hydrogen photoproduction[J].International Journal of Hydrogen Energy,2013,38(16):6593-6599.
(责任编辑 薛 荣)
Preparation and photocatalytic properties of ZnFe2O4/polyaniline composites
RAN Fang, MAO Weixing, ZHAO Qiaoyun, GAO Xiaoyu, SHEN Xinyi,TONG Guoxiu, LÜ Tianxi, GONG Peijun
(CollegeofChemistryandLifeSciences,ZhejiangNormalUniversity,Jinhua321004,China)
Zinc ferrite/polyaniline composites (ZnFe2O4/PANI) with different mass ratios were prepared via chemically oxidative polymerization method. The composites were then characterized and their adsorption performance and the visible light photocatalytic activity were evaluated by using Rhodamine B (RhB) as the model organic pollutant. The results showed that the composites had an enlarged mass percent of PANI with gradually increased HCl doping degree, improved conductivity and absorption capacity for visible light as the mass ratio of ZnFe2O4to aniline (wZ/A) decreased. Meanwhile, modification of ZnFe2O4with PANI promoted the adsorption of RhB on the catalyst. The equilibrium adsorption capacity for RhB was gently elevated with the decreasingwZ/A. However, the photocatalytic activity of ZnFe2O4/PANI initially increased and then decreased. The photocatalyst withwZ/A=10% exhibited the best photocatalytic activity under visible light irradiation, excellent stability in five cycles and good magnetic responsiveness for recollection. A possible photocatalytic mechanism was proposed according to the trapping experiment of active spices.
zinc ferrite; polyaniline; chemically oxidative polymerization; photocatalysis; magnetic responsiveness
10.16218/j.issn.1001-5051.2017.01.010
2016-06-14;
2016-09-12
浙江省公益技术应用研究项目(2016C31014;2015C31022);国家级大学生创新创业训练计划项目(201510345020);国家留学基金资助项目
冉 方(1990-),女,河南郑州人,硕士研究生.研究方向:纳米复合材料.
宫培军.E-mail: skygpj@zjnu.cn
O643.36
A
1001-5051(2017)01-0064-07
