基于CFS算法研究肾癌中的关键基因
2017-07-06张梦莹上海大学生命科学学院上海200444
张梦莹,卢 易,钮 冰,苏 强 (上海大学生命科学学院,上海 200444)
·基础与转化医学·
基于CFS算法研究肾癌中的关键基因
张梦莹,卢 易,钮 冰,苏 强 (上海大学生命科学学院,上海 200444)
目的:识别肾细胞癌(RCC)中的关键基因,并揭示其在肿瘤中的作用机理.方法:从GEO数据库下载芯片数据GSE53757,筛选它们之间的差异表达基因(DEGs).使用DAVID在线工具对DEGs进行GO功能注释和KEGG富集分析,然后使用基于特征子集相关性(CFS)的变量筛选方法筛选DEGs的关键基因,并根据筛选出的关键基因,使用支持向量机(SVM)方法建立筛选RCC样本和正常对照样本的分类预测模型.结果:共筛选到541个DEGs,包括312个上调基因和229个下调基因.选择21个作为特征基因,通过SVM方法建立RCC样本和正常对照样本之间的分类模型,其预测精度为97.2%.此外,STRING数据库筛选的Top10Hub基因中也发现了4个与CFS算法筛选出的特征基因重合的Hub基因(CD40,EGFR,CAV1和TGFA).结论:CFS是用于筛选RCC中关键基因的有用工具.并且,CD40,EGFR,CAV1和TGFA这4个基因很可能为诊断RCC的目标基因.
基因表达谱;癌症分类;基因选择;肾癌;CFS算法
0 引言
肾细胞癌(renal cell carcinoma,RCC)是成人肾脏中最常见的一种恶性肿瘤,约占成人恶性肿瘤的3%[1].2012年,美国的患RCC的人数超过64 770人,且有13 570例患者死于该疾病[2].且RCC对放射治疗和化疗的抵抗力较强,为此需要开发新的治疗策略和药物.
在本研究中,首先筛选RCC样本和正常对照样本之间的差异表达基因(differentially expressed genes,DEGs),使用DAVID对DEGs进行GO功能注释和KEGG富集分析.然后,使用基于特征子集相关性(correlation⁃based feature subset,CFS)的变量筛选方法筛选 DEGs的关键基因,使用支持向量机(support vector machine,SVM)建立RCC样本和正常对照样本之间的分类模型.
1 材料和方法
1.1 芯片数据来源 从美国国立生物技术信息中心(National Center of Biotechnology Information,NCBI)的基因表达谱数据库(gene expression omnibus,GEO)下载芯片数据GSE53757,芯片平台为GPL570([HGU133Plus2]Affymetrix Human Genome U133 Plus 2.0 Array),下载该芯片数据和注释文件,选取其中的72例RCC样本和72例正常对照样本进行分析.
1.2 数据预处理与差异基因的计算 获得原始数据后,使用R软件分别对两组数据进行分析,用Affy包中的 GCRMA(GeneChip robust multiarray average)法[3]对芯片数据进行归一化处理,获得标准化后的芯片表达谱,然后使用线性回归模型软件包Limma包中的 T⁃test法[4]计算两组数据中的 DEGs,选取|logFC|>2.0和P<0.05作为显著性阈值,从而得到疾病组的差异表达情况.
1.3 基因本体和通路富集分析 基因本体(geneontology,GO)[5-6]是一种用于注释所有基因及基因产物特征生物学属性的一种有效方法.KEGG(Kyoto encyclopedia of genes and genomes)[7-8]是将基因、基因组信息以及更高层次的功能信息结合起来进行系统化分析的数据库.在本研究中,使用DAVID在线工具[9]对DEGs进行GO富集和KEGG通路富集分析,以P<0.05作为显著性阈值,从而得到DEGs参与的主要功能信息和通路信息.
1.4 蛋白质⁃蛋白质相互作用(PPI)网络构建STRING数据库[10-11]是用于评估蛋白质⁃蛋白质相互作用(protein⁃protein interaction,PPI)的一种在线工具.为了评估DEGs之间的相互作用关系,DEGs被上传到STRING数据库中进行计算,在STRING的计算结果中选取相互作用得分在0.4以上的基因作为显著性基因.
1.5 CFS的变量筛选方法 CFS变量筛选方法[12-13]的原理是在进行变量筛选时,主要考虑特征之间的相关性.通过计算特征子集中全部特征对于分类的联合贡献来判断特征子集的类间辨别能力大小,不再仅考虑单个特征对于分类的贡献.将与类的相关性很低的不相关的特征忽略,将与一个或多个特征高度相关的冗余特征去掉.特征的选取将仅仅取决于其在实例空间中与其他特征相关的程度.
1.6 SVM SVM[14-15]是Vapnik等于1995年首先提出的一种新型的机器学习方法.SVM将向量映射到一个更高维的空间里,在这个空间里建立有一个最大间隔超平面,在分开数据的超平面的两边各有两个互相平行的超平面.SVM算法建立方向合适的分类超平面,使两个与之平行的超平面间的距离最大化,使得所属不同类别的样本有一个尽可能大的明显的差距.新的样本实例则映射到相同的空间中,并基于它们落在所属间隙的哪一侧来预测属于哪一个类别.

图1 DEGs的GO富集分析,纵坐标为GO term
2 结果
2.1 DEGs的筛选 通过使用R软体中的Limma包来筛选RCC样本和正常对照样本之间的DEGs,以|logFC|>2.0及P<0.05为显著性阈值,从而筛选出541个DEGs,其中包括312个上调基因和229个下调基因.
2.2 DEGs的GO富集分析 如图1所示,GO分析分别针对上调基因和下调基因的3个不同方面(即生物过程(biological process,BP),细胞组分(cellular component,CC)和分子功能(molecular function,MF))进行分析.选择P<0.05作为显著性的阈值,并按照富集倍数或富集得分(⁃log 10(P⁃value))对结果进行排序.结果显示,在所有比较组中,有459个BP、54个CC和116个MF上调(P<0.05),同时,有715 BP、53个CC和67个MF下调(P<0.05).根据倍数富集或富集得分排列的按BP,CC和MF分类的所有比较组中的前10个一般改变的GO项见图1.
2.3 DEGs的KEGG途径分析 KEGG富集分析的结果见表1,上调的DEGs在代谢途径、抗生素生物合成、缬氨酸、亮氨酸和异亮氨酸的降解、收集管道酸分泌和碳代谢等Pathway显著富集,而下调的DEGs在病毒性心肌炎、HIF⁃1信号通路、粘着斑、吞噬体和用于IgA生产肠免疫网络等pathway显著富集.
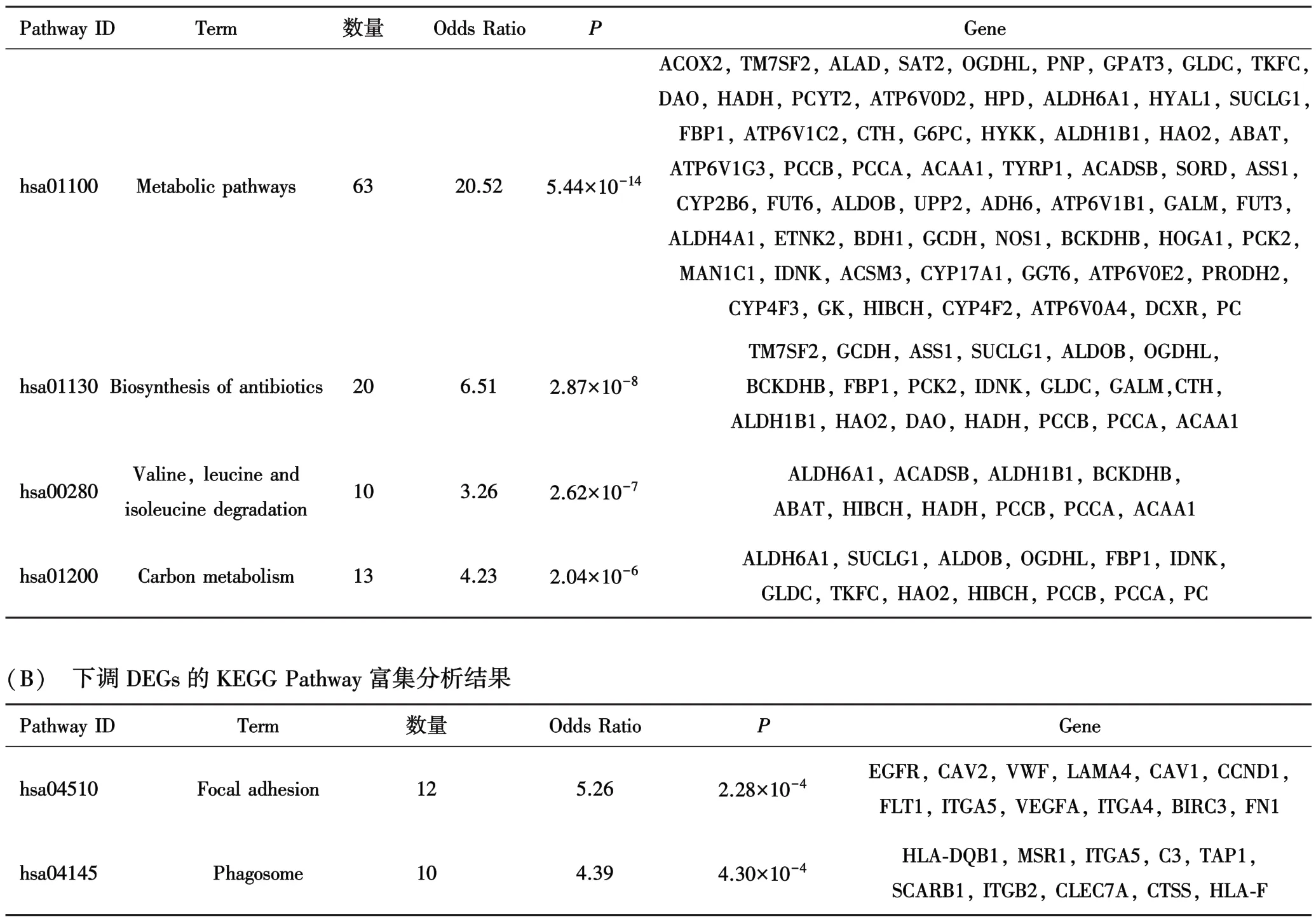
表1 DEGs的KEGG Pathway富集分析结果(A) 上调DEGs的KEGG Pathway富集分析结果
2.4 使用CFS算法筛选特征基因 采用CFS方法筛选特征基因,通过计算共筛选出21个基因(UMOD,KNG1,CALB1,SLC12A1,ALB,DIO1,SLC22A8,FABP1,HPD,ALDOB,KCNJ1,NPHS2,XPNPEP2,CLCNKB,SLC13A3,TMEM52B,CD40,EGFR,CAV1,ABAT和TGFA),并基于筛选出的基因建立分类预测模型.
2.5 癌症样本和正常对照样本的分类 基于上述结果,使用SVM建立疾病组与正常组的分类模型.将SVM中的核函数设置为ε=0.01,C=1,然后使用留一交叉验证法(leave⁃one⁃out cross⁃validation,LOOCV)进行验证[16],评估该模型的准确度.留一法结果显示该分类模型的预测准确度为97.2%.
3 讨论
3.1 CFS方法与其他变量筛选方法的比较 将CFS算法的筛选结果与其他 3种基因筛选算法(Re⁃liefF[17-18],mRMR[19-20]和 Random Forest[21-22])进行比较,结果见表2,CFS算法在这4种特征变量筛选方法中,筛选的特征基因数量最少且预测准确度最高.

表2 mRMR,ReliefF,Random Forest和CFS 4种算法的比较
3.2 通过PPI网络筛选的Hub基因 基于STRING数据库中的信息,我们筛选出得分最高的Top10 Hub基因(CD40,ATP6V0D2,EGFR,NPHS1,FYB,CAV1,VEGFA,PCCB,TGFA和 KLHL3),在这些Hub基因中,包含有4个 CFS筛选出的特征基因(CD40,EGFR,CAV1和TGFA).
CD40是参与免疫调节的细胞表面受体,其配体在活化的T细胞上表达[23].CD40在肾脏中的作用可能是增强细胞因子和趋化因子的产生[24].
EGFR(epidermal growth factor receptor)自分泌途径在许多癌症发展过程中发挥着重要作用,包括细胞增殖,细胞凋亡,血管生成和转移扩散[25].
Cav1(Caveolin⁃1)是caveolae的主要结构蛋白,在胞吞、细胞转运、细胞信号转导中发挥着重要作用[26].
TGFA(Transforming growth factor⁃α)[27]被认为是EGF/TGF⁃α受体的配体,因为它在早期发育中广泛存在于各种胚胎组织中[28].TGFA可能是控制发育过程的关键分子,会影响原始肾细胞的早期分化和生长以及成人肾细胞中肿瘤的转化.本研究DEGs分析的结果表明,上述4个基因在肿瘤样本中均出现明显的下调趋势.
根据前面的结果进行分析,CD40,EGFR,CAV1和TGFA很可能是诊断RCC的靶基因,因为它们在分类预测模型和PPI网络中都起到了重要作用.
4 结论
本研究中,CFS基因筛选方法提供了一种对DEGs生物信息学分析结果进行进一步分析的方法,可以筛选出RCC中的关键基因.此外,计算结果还表明CD40,EGFR,CAV1和TGFA 4个基因可能为诊断RCC的目标基因.
[1]Ouzaid I.Kidney cancer[J].Prog Urol,2015,25:85.
[2]Stewart B,Wild C.World cancer report 2014[M].Int Agency Res Cancer,2014.
[3]Aoki Y,Watanabe T,Saito Y,et al.Identification of CD34+and CD34-leukemia⁃initiating cells in MLL⁃rearranged human acute lym⁃phoblastic leukemia[J].Blood,2015,125(6):967-980.
[4]Mallik S,Bhadra T,Maulik U.Identifying epigenetic biomarkers using maximal relevance and minimal redundancy based feature selec⁃tion for multi⁃omics data[J].IEEE Trans Nanobiosci,2017,16(1):3-10.
[5]Harris MA,Clark J,Ireland A,et al.The gene ontology(GO)database and informatics resource[J].Nucleic Acids Res,2004,32:D258-D261.
[6]Blake JA,Dolan M,Drabkin H,et al.Gene ontology annotations and resources[J].Nucleic Acids Res,2013,41(D1):530-535.
[7]Richter S,Fetzer I,Thullner M,et al.Towards rule⁃based metabolic databases:a requirement analysis based on KEGG[J].Int J Data Min Bioinform,2015,13(3):289-319.
[8]Maiorano F,Ambrosino L,Guarracino MR.The MetaboX library:building metabolic networks from KEGG database[J].Quantitative Biol,2014.
[9]Jiao X,Sherman BT,Huang da W,et al.DAVID⁃WS:a stateful web service to facilitate gene/protein list analysis[J].Bioinformatics,2012,28(13):1805-1806.
[10]Szklarczyk D,Franceschini A,Kuhn M,et al.The STRING database in 2011:functional interaction networks of proteins,globally integrat⁃ed and scored[J].Nucleic Acids Res,2011,39(Database issue):D561-D568.
[11]Szklarczyk D,Franceschini A,Wyder S,et al.STRING v10:pro⁃tein⁃protein interaction networks,integrated over the tree of life[J].Nucleic Acids Res,2015,43(D1):D447-D452.
[12]Senliol B,Gulgezen G,Yu L,et al.Fast correlation based filter(FCBF)with a different search strategy[M].https://doi.org/10.1109/ISCIS.2008.4717949.
[13]Cataltepe Z,Uluyaĝmur M,Tayfur E.Feature selection for movie recommendation[J].Turkish J Electric Eng&Computer Sci,2016,24(3):833-848.
[14]Ben⁃Hur A,Horn D,Siegelmann HT,et al.Support vector clustering[J].J Mach Learn Res,2002,2(2):125-137.
[15]Cortes C,Vapnik V.Support⁃Vector Networks[J].Mach Learning,1995,20(3):273-297.
[16]Kohavi R.A study of cross⁃validation and bootstrap for accuracy esti⁃mation and model selection[J].Int joint conference artificial intelli⁃gence,1995:1137-1143.
[17]Robnik⁃Šikonja M,Kononenko I.Theoretical and empirical analysis of ReliefF and RReliefF[J].Mach Learning,2003,53(1):23-69.
[18]Xue ZY,Liu XQ.Feature selection method for object⁃oriented build⁃ing targets recognition based on ReliefF,GA and SVM[J].Eng Sur⁃veying&Mapping,2017.
[19]Peng H,Long F,Ding C.Feature selection based on mutual information:criteria of max⁃dependency,max⁃relevance,and min⁃redundancy[J].IEEE Transactions on Pattern Analysis and Machine Intelligence,2005,27(8):1226-1238.
[20]Li BQ,Zheng LL,Feng KY,et al.Prediction of linear B⁃cell epitopes with mRMR feature selection and analysis[J].Curr Bioin⁃form,2016,11(1):22-31.
[21]Díaz⁃Uriarte R,Alvarez de Andrés S.Gene Selection and Classifica⁃tion of Microarray Data Using Random Forest[J].BMC Bioinformat⁃ics,2006,7:3.
[22]Pashaei E,Ozen M,Aydin N.Gene selection and classification approach for microarray data based on Random Forest Ranking and BBHA[C]//IEEE⁃EMBS International Conference on Biomedical and Health Informatics.IEEE,2016:308-311.
[23]Byrne KT,Vonderheide RH.CD40 stimulation obviates innate sensors and drives T cell immunity in cancer[J].Cell Rep,2016,15(12):2719-2732.
[24]Woltman AM,de Haij S,Boonstra JG,et al.Interleukin⁃17 and CD40⁃ligand synergistically enhance cytokine and chemokine produc⁃tion by renal epithelial cells[J].J Am Soc Nephrol,2000,11(11):2044-2055.
[25]Bartholomew C,Eastlake L,Dunn P,et al.EGFR targeted therapy in lung cancer;an evolving story[J].Respir Med Case Rep,2017,20:137-140.
[26]Qin L,Zhu N,Ao BX,et al.Caveolae and Caveolin⁃1 Integrate Re⁃verse Cholesterol Transport and Inflammation in Atherosclerosis[J].Int J Mol Sci,2016,17(3):429.
[27]Junaid M,Narayanan MB,Jayanthi D,et al.Association between maternal exposure to tobacco,presence of TGFA gene,and the occur⁃rence of oral clefts.A case control study[J].Clin Oral Investig,2017.
[28]Mattii L,Bianchi F,Da Prato I,et al.Renal cell cultures for the study of growth factor interactions underlying kidney organogenesis[J].In Vitro Cell Dev Biol Anim,2001,37(4):251-258.
Identification of key genes in renal cell carci⁃noma based on CFS gene selection algorithm
ZHANG Meng⁃Ying,LU Yi,NIU Bing,SU Qiang College of Life Science,Shanghai University,Shanghai 200444,China
AIM:To identify key genes signatures in renal cell carcinoma(RCC) and uncover their potentialmechanisms.METHODS:Firstly,the gene expression profiles of GSE53757,which contained 144 samples,including 72 RCC samples and 72 controls,was downloaded from GEO database.And then differen⁃tially expressed genes(DEGs)between the RCC samples and the controls were identified.After that,GO and KEGG enrichment analyses of DEGs were performed by DAVID.Furthermore,corre⁃lation⁃based feature subset(CFS)method was applied to the se⁃lection of key genes of DEGs.In addition,the classification model between the RCC samples and the controls was built by support vector machines(SVM) based on selection of key genes.RESULTS:DEGs contained 541 genes,including 312 up⁃regu⁃lated and 229 down⁃regulated genes.A total of 21 DEGs were selected as the feature genes to build the classification model between the RCC samples and the controls by CFS method.The accuracy of the classification model is 97.2%.Besides,four feature genes(CD40,EGFR,CAV1 and TGFA)also can been found in the top 10 hub genes screened by STRING database.CONCLUSION:It indicats that CFS is a useful tool to identify key genes in RCC.Besides,we also predicts genes such as CD40,EGFR,CAV1 and TGFA might be target genes for diagno⁃sing the RCC.
gene expression profiles; cancer classification;gene selection;renal cell carcinoma;CFS
2095⁃6894(2017)06⁃16⁃04
R737.11
A
2017-04-05;接受日期:2017-04-20
张梦莹.E⁃mail:18800208364@163.com
苏 强.博士,研究员.研究方向:生物信息学.E⁃mail:su@shu.edu.cn
