主动脉结合性质导致药物副作用的研究进展
2017-05-10冉兆晋柴宝山
冉兆晋,柴宝山
(沈阳化工研究院有限公司医药研究室,辽宁沈阳 110021)
主动脉结合指具有特定结构的化合物与原弹性蛋白中的醛基赖氨酸发生反应,进而破坏动脉弹性蛋白的形成乃至降解的性质。研究发现,莫唑胺[1]、罗非昔布[2-4]和AZD5248[5]等(图1)已上市或处于临床前研究的药物具有主动脉结合性质,导致药物副作用,尤其增加了心血管疾病(cardiovascular disease,CVD)的风险。本文对具有主动脉结合性质的药物进行梳理总结,分析其产生的机制,总结相关化合物的结构特点,并对其体内外研究方法进行总结,有助于药物研发工作者关注该性质,规避药物设计中可能出现的风险,为后续相关研究提供方法和思路。
1 主动脉结合发生机制
原弹性蛋白中的赖氨酸残基通过赖氨酰氧化酶(lysyl oxidase,LOX)被氧化成醛基赖氨酸,醛基赖氨酸与赖氨酸残基可以4-4交联形成锁链素与异锁链素,原弹性蛋白通过锁链素与异锁链素共价交联形成正常的弹性蛋白(图2)[2-3]。而具有主动脉结合性质的化合物可与原弹性蛋白中的醛基共价结合,破坏锁链素与异锁链素的形成,进而破坏弹性蛋白的正常形成(图3)[2-5]。弹性蛋白具有非常缓慢的周转率[6-7],化合物以剂量大小和给药频率在主动脉中逐渐累积,进而破坏弹性蛋白形成,甚至引起降解,主动脉出现功能障碍,最后导致CVD。
2 具有主动脉结合性质的药物
2.1 肼屈嗪(hydralazine)
肼屈嗪是用于治疗肾型高血压的药物,是最早被发现具有主动脉结合性质的药物。在早期研究中,Moore-Jones等[8]给小鼠注射[14C]标记的肼屈嗪,通过放射自显影技术发现,小鼠主动脉上的放射性物质含量远高于其他组织,他们认为肼屈嗪这种对血管的亲和力与其毒性相关,推测该药物可能导致血管损伤与红斑狼疮。该研究为第一次公开报道药物的主动脉结合性质,但作者并未对该结合机制进行推测或深入研究。随后,肼屈嗪因较大的副作用被FDA警告,禁止用于冠状动脉病变和脑血管硬化等治疗。
根据现在阐明的主动脉结合发生机制,推测肼屈嗪与主动脉壁的结合是肼屈嗪上的肼基与原弹性蛋白中醛基赖氨酸的醛基发生反应生成了碳氮双键(图4),导致肼屈嗪富集于主动脉壁上难以正常代谢,导致血管病变,进而增加了CVD发病的概率。
2.2 咪唑(imidazole)
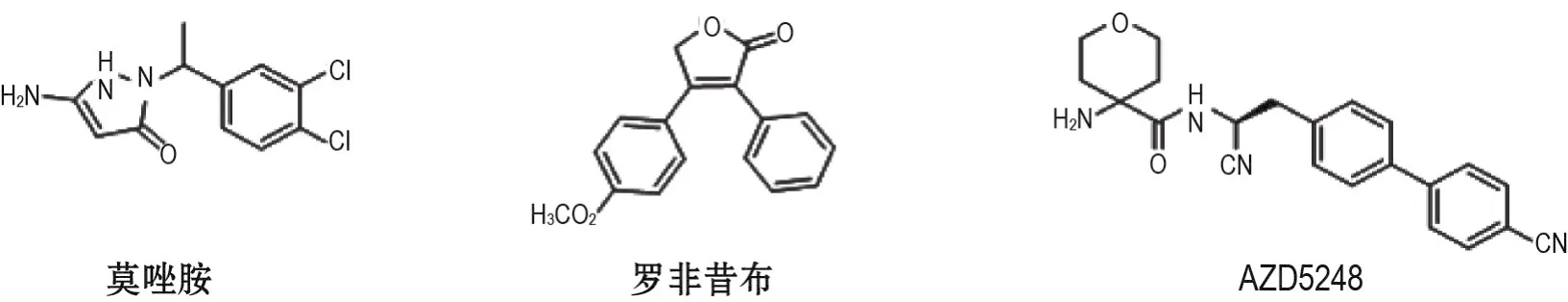
图1 具有主动脉结合性质的部分药物.
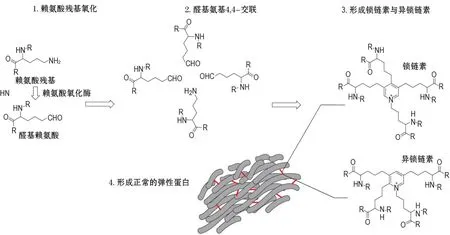
图2 正常弹性蛋白的形成过程.
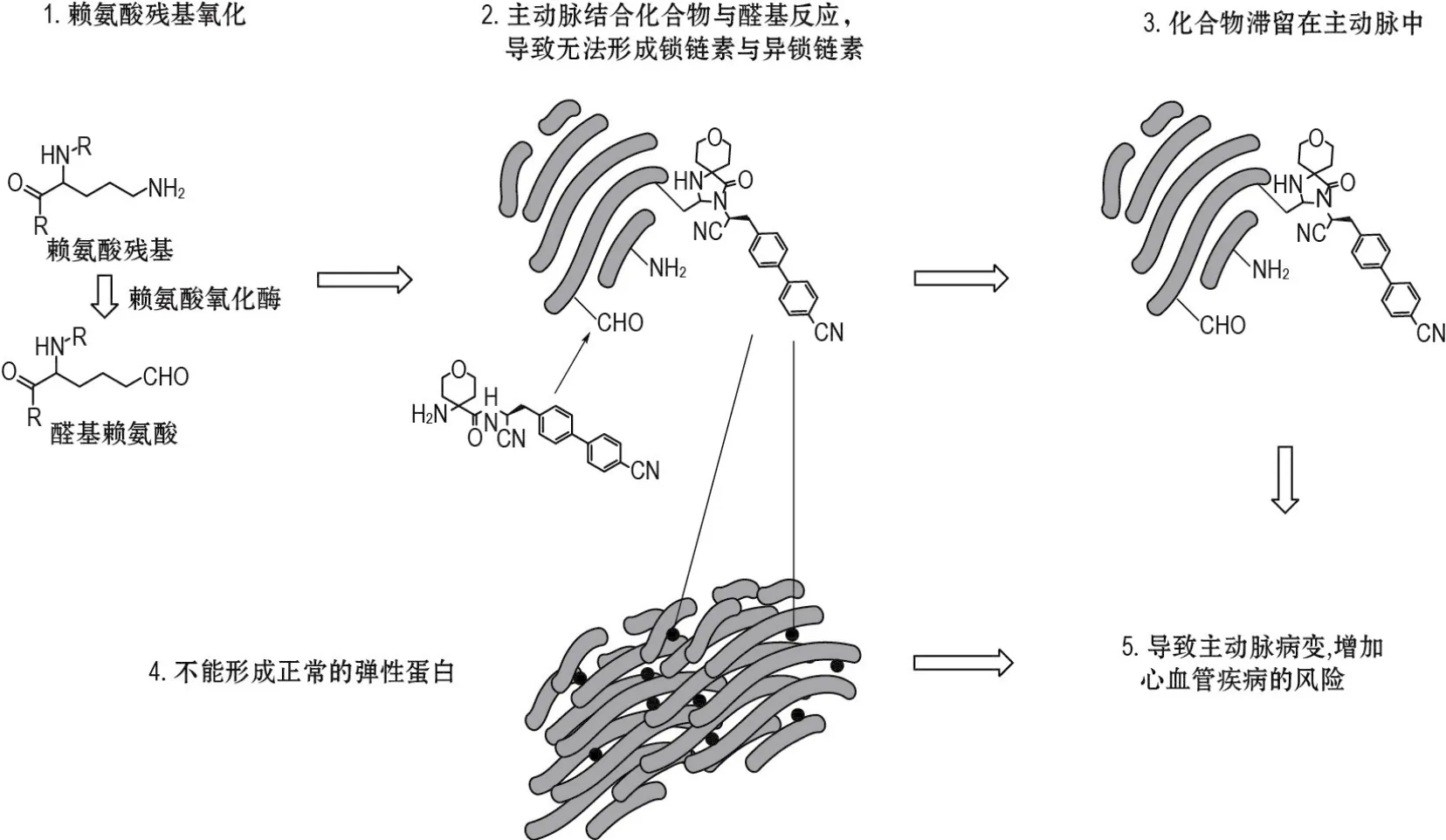
图3 受主动脉结合药物干扰的弹性蛋白病变过程.
咪唑作为重要的化工中间体大量应用于精细化学品合成与树脂固化剂和黏合剂中。Ohta等[9-12]对咪唑类化合物的主动脉结合性质进行了一系列研究。大鼠静脉注射给予[14C]咪唑类化合物(3 μmol·kg-1)后,通过放射自显影确认不可逆结合位点主要位于主动脉的弹性蛋白。但进一步的体外实验却发现,[14C]咪唑类化合物与主动脉(犬动脉切片)不能结合。体内实验发现,普罗地芬(proadifen)可增加[14C]咪唑类化合物主动脉结合,推测是细胞色素P450酶催化咪唑产生的代谢物与主动脉发生了结合[9]。为进行进一步的机制研究,Ohta等[10]在体外实验中采用氯化铜加维生素C处理的[14C]咪唑化合物,发现其与犬动脉切片发生主动脉结合,而用还原物质硼氢化钠等预处理犬动脉切片可减少主动脉结合,据此推测是咪唑的氧化代谢物与弹性蛋白中的醛基发生了键合。Ohta等[11]进一步采用咪唑氧化代谢物咪唑啉酮与醛基化合物在体外模拟主动脉结合的过程。结果表明,其可能是通过羟醛缩合反应发生结合。而且通过大鼠静脉注射[14C]咪唑啉酮化合物实验证实了体外实验的结果,认为细胞色素P450酶催化产生的咪唑啉酮代谢物是主动脉结合的原因[12](图5)。一系列体内与体外研究基本阐明了主动脉结合发生的原因和可能的结合机制,为后续的相关研究打下了良好的基础。
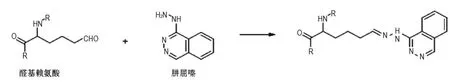
图4 肼屈嗪的主动脉结合机制.肼屈嗪上的肼基与原弹性蛋白中醛基赖氨酸的醛基发生反应生成了碳氮双键.
2.3 莫唑胺(muzolimine)
莫唑胺是治疗高血压的药物,因严重副作用而退市。Schmidt等[1]在早期研究中发现,其除了较大的神经副作用外,还具有主动脉结合的性质。大鼠喂食[14C]莫唑胺(3 mg)2周后,其动脉的放射活性是皮肤和肌肉的60~300倍,给药剂量的0.04%结合在了主动脉弹性蛋白上。该研究还建立了用胰弹性蛋白酶水解主动脉释放结合的莫唑胺的方法,为体外定量检测主动脉结合化合物奠定基础。该研究还使用[4,5-3H]赖氨酸标记大鼠弹性蛋白,莫唑胺处理2周,发现弹性蛋白中锁链素与异锁链素显著低于空白对照。表明其机制可能是干扰了锁链素与异锁链素的形成而影响动脉(图6)。
2.4 罗非昔布(rofecoxib)
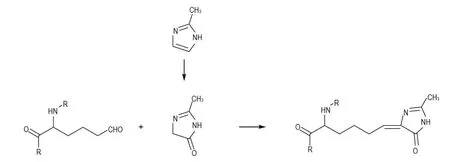
图51 -甲基咪唑的主动脉结合机制.咪唑类化合物在P450酶的作用下氧化生成咪唑啉酮,咪唑啉酮和醛基赖氨酸发生羟醛缩合反应生成碳碳双键,发生不可逆结合.
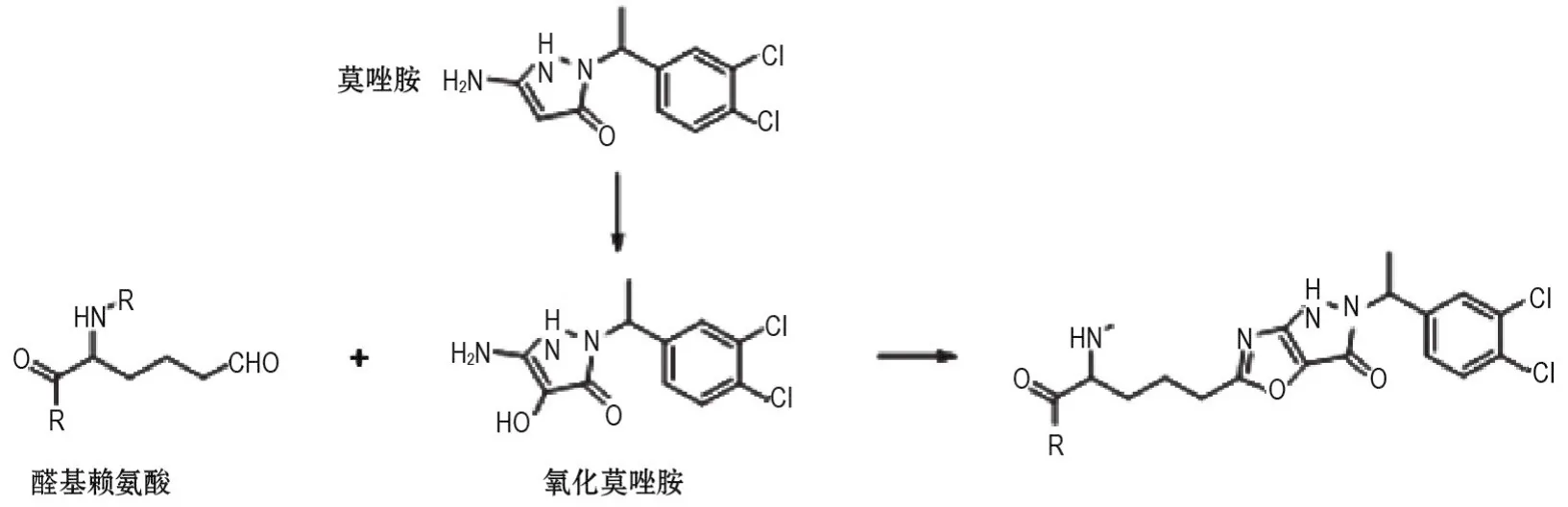
图6 莫唑胺的主动脉结合机制.莫唑胺先在体内被氧化为氧化莫唑胺,再与醛基赖氨酸的醛基脱水关闭噁唑环,发生不可逆结合.
罗非昔布为环氧化酶2(cyclo-oxygenase-2,COX-2)特异性抑制剂,具有很强的止痛和抗炎作用。2000年,发现其具有较大的副作用[13];2001年,美国FDA因其副作用对其进行了警告[14];2004年,由于确切的心血管副作用,默沙东主动召回退市[15],导致了巨大的经济损失。Otitate等[2-3]对罗非昔布的副作用进行了系统研究,用COX-2抑制剂[14C]罗非昔布、[14C]塞来昔布和[14C]CS-706处理大鼠(均为2 mg·kg-1),全身放射自显影技术与定量组织分析显示,只有罗非昔布在大鼠动脉中有集聚;进一步的分析显示,其主要结合在弹性蛋白上。该研究还第一次使用了斑马鱼模型进行主动脉结合的全身放射自显影技术,直观地观察到[14C]罗非昔布的集聚现象[2]。进一步研究表明,罗非昔布是直接与主动脉产生结合,而不是其代谢产物5-羟基罗非昔布(图7)[3]。除罗非昔布外,其他COX-2抑制剂不与主动脉结合,而罗非昔布也不与动脉内皮细胞结合,而是特异性结合于弹性蛋白中。此外,使用苯甲醛替代动脉切片的方法进行体外结合实验,发现罗非昔布可与苯甲醛在体外产生结合[2-3]。一系列研究表明,罗非昔布相对于其他COX-2抑制剂,具有主动脉结合的性质,可导致更高的心血管发病概率[2-4]。主动脉结合性质是罗非昔布退市的主要原因。
2.5 AZD5248
AZD5248是阿斯利康(AstraZeneca)公司开发的治疗慢性阻塞性肺病的临床前候选药物,由于早期毒理研究发现其具有主动脉结合性质(图8)[5],不得不放弃该分子骨架,重新设计开发了AZD7986(图9)[16],开发时间虽耽误并付出了更多的研发经费,但其从根源上避免了后期的心血管副作用,大大降低了该药物的后期市场风险。在研究过程中使用了早期研究者建立的一系列体内和体外研究方法,并且完善了相关的体外模拟研究方法,如采用丙醛与苯甲醛共同进行体外模拟实验,避免单独使用苯甲醛可能带来的假象。还意外地发现了AZD5248类似物卡普瑞林(campromorelin)的主动脉结合假阳性,并开发了体外假阳性研究方法。
2.6 ZD4407
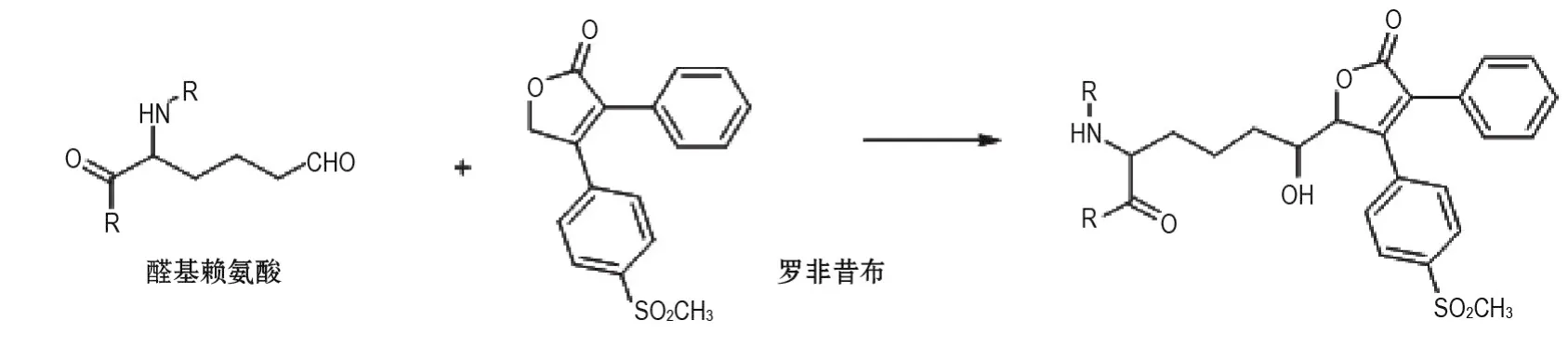
图7 罗非昔布的主动脉结合机制.通过体外实验结果推测得出,罗非昔布与醛基赖氨酸的醛基加成,形成共价不可逆结合.
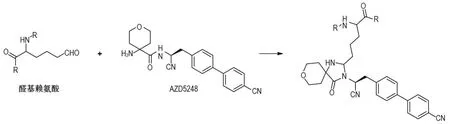
图8 AZD5248的主动脉结合机制.AZD5248通过其结构中的氨基乙酰胺结构与醛基赖氨酸的醛基关咪唑啉酮环发生不可逆结合.
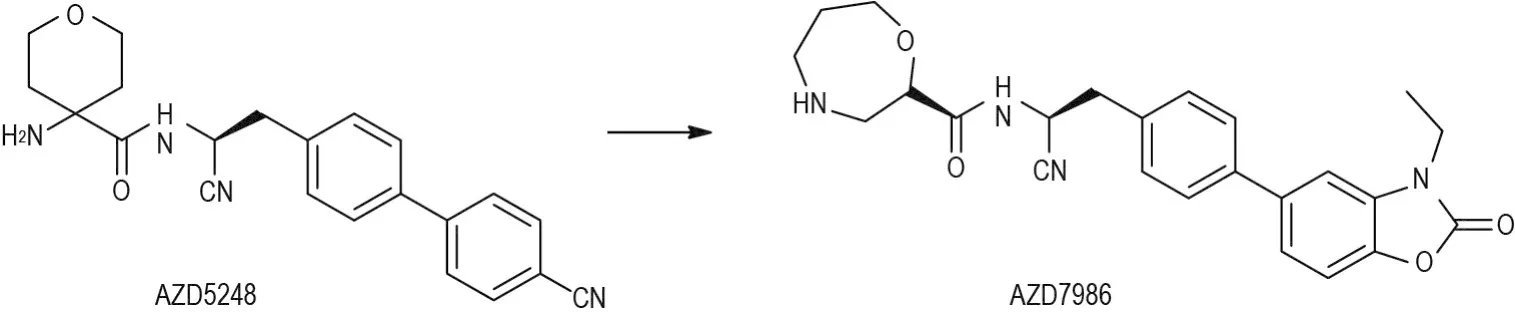
图9 AZD7986由AZD5248改造获得.AZD7986无氨基乙酰胺结构,在保持活性的基础上避免了主动脉结合的副作用.
ZD4407是一个处于开发中的5-脂氧化酶抑制剂。毒理研究发现,其也具有潜在的主动脉结合性质(图10)[5]。因此,对其进行相关的结构改造可避免主动脉结合导致的副作用。
3 主动脉结合研究方法
3.1 体内研究
3.1.1 [14C]标记化合物定量分析
将[14C]标记化合物(用于主动脉中的放射性测量)溶解,ig给予实验动物,一定时间后安乐死,收集胸主动脉。使用不同的蛋白水解酶分解主动脉的各个成分并提取,处理后使用超高效液相色谱对比定性定量分析[2-5]。该研究方法需要模型动物、放射标记化合物和带有放射性检测仪的HPLC等设备。
3.1.2 [14C]标记化合物定量全身放射自显影技术分析
定量全身放射自显影技术(quantitative whole body autoradiography,QWBA),利用[14C]标记化合物在大鼠或斑马鱼体内组织分布进行研究。这是现在最有效直观的研究方法[4-5]。实验动物给予不同剂量[14C]标记化合物一段时间后,分别于不同时间进行处理并冷冻,处理后使用磷屏曝光,将激发后的磷屏在同位素影像分析仪中读取信号,呈现放射性化合物的动物组织分布图像,并可利用其灰度值对动物体内放射性化合物进行相对定量。在主动脉结合研究中,斑马鱼被更多地用于该实验研究。随着时间的延长,放射标记化合物在斑马鱼的主动脉中表现出明显的结合留驻现象。该研究方法需要模型动物、放射标记化合物和同位素影像分析仪等设备。
3.2 体外研究
3.2.1 体外苯甲醛或丙醛结合模拟分析
使用苯甲醛或丙醛作为原弹性蛋白中醛基赖氨酸的模拟化合物,与检测目标化合物混合反应一段时间,24 h后使用液质联用检测[2,5],模拟检测机制(图10)。利用丙醛与苯甲醛的醛基与AZD5248的氨基乙酰胺结构反应,生成咪唑啉酮环。该研究方法需要液质联用设备。
3.2.2 体外[14C]标记化合物竞争性结合分析
将大鼠动脉剥离并匀浆后与检测目标化合物一起孵育一段时间,随后加入有放射性标记的确定具有动脉结合性质的模式化合物,再次孵育一段时间,分离提取检测目标溶液,使用多用途闪烁计数器对其进行放射性检测来判断结合情况,该法的特点是可体外判断出一些假阳性化合物[3-5]。
卡普瑞林(生长激素释放刺激肽类药)与具有主动脉结合活性的AZD5248有相似的α-氨基酰胺结构(图11),推测其同样具有主动脉结合活性。体外苯甲醛或丙醛结合模拟分析为阳性(图12);但竞争性结合实验为阴性(图13),与QWBA分析结果一致,说明在假阳性判断上,竞争性结合分析较普通的苯甲醛丙醛结合分析具有优势。推测卡普瑞林具有可逆的弱结合,在竞争化合物AZD5248的干扰下会解离。导致该结果的深层次的机制与原因还有待进一步研究[5]。该研究方法需要模型动物、放射标记化合物和多用途闪烁计数器等设备。
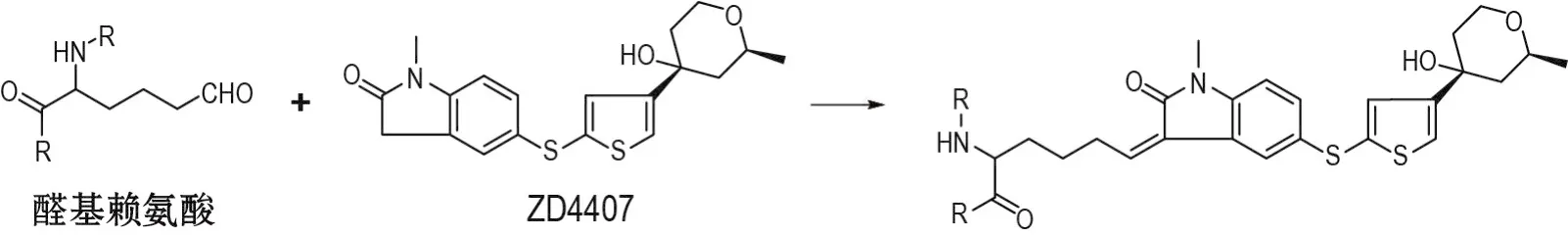
图10 ZD4407的主动脉结合机制.ZD4407中的二氢吲哚-2-酮与醛基赖氨酸的醛基发生缩合反应,形成不可逆结合.
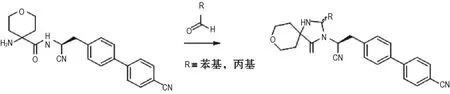
图11 AZD5248模拟检测反应机制.
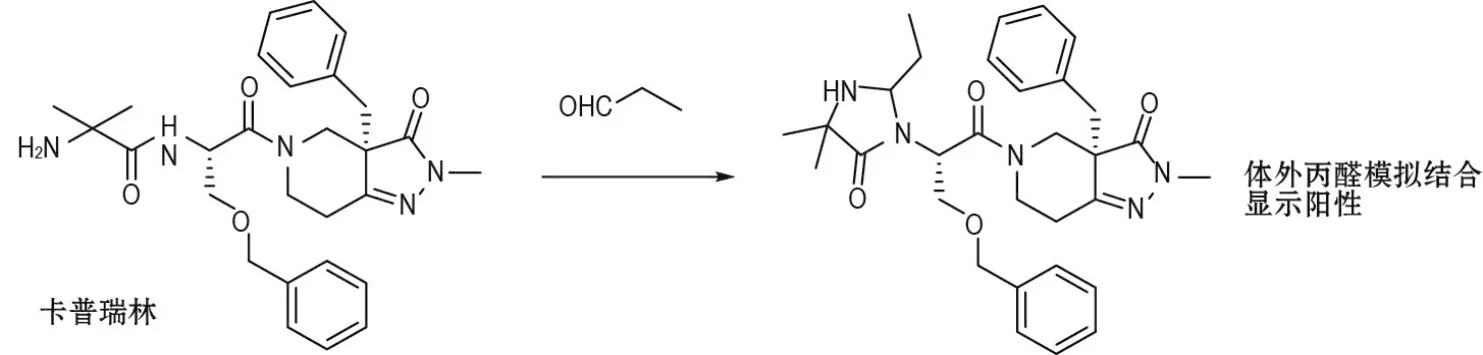
图12 卡普瑞林体外丙醛模拟结合.
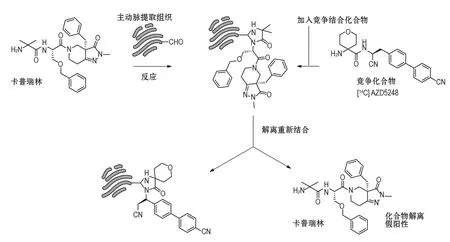
图13 体外[14C]标记AZD5248与卡普瑞林主动脉竞争性结合.
4 结语
药物因主动脉结合导致副作用的相关研究报道虽较少,但从其产生的机制上分析,尚可推测出许多药物的化学结构具有潜在的主动脉结合的可能性。阿斯利康的AZD5248不会是最后一个因为主动脉结合导致的副作用而重新研发的临床候选药物。中国的创新药物研发应关注并了解主动脉结合的基本原理,熟悉发生主动脉结合的结构,在药物设计过程中进行早期规避,降低药物在临床乃至上市后的CVD风险。本综述对广大药物研发工作者具有一定的现实参考意义。
参考文献:
[1] Schmidt A,Busse WD,Garthoff B,Gau W,Ritter W,Wünsche C,et al.Influence of muzolimine on arterial wall elastin[J].Biochem Pharmacol,1984,33(12):1915-1921.
[2] Oitate M,Hirota T,Murai T,Miura S,Ikeda T.Covalent binding of rofecoxib,but not other cyclooxygenase-2 inhibitors,to allysine aldehyde in elastin of human aorta[J].Drug Metab Dispos,2007,35(10):1846-1852.
[3] Oitate M,Hirota T,Takahashi M,Murai T,Miura S,Senoo A,et al.Mechanism for covalent binding of rofecoxib to elastin of rat aorta[J].J Pharmacol Exp Ther,2007,320(3):1195-1203.
[4] Oitate M,Hirota T,Koyama K,Inoue S,Kawai K,Ikeda T.Covalent binding of radioactivity from[14C]rofecoxib,but not[14C]celecoxib or[14C]CS-706,to the arterial elastin of rats[J].Drug Metab Dispos,2006,34(8):1417-1422.
[5] Bragg RA, BrocklehurstS, Gustafsson F,Goodman J,Hickling K,MacFaul PA,et al.Aortic binding of AZD5248:mechanistic insight and reactivity assays to support lead optimization[J].Chem Res Toxicol,2015,28(10):1991-1999.
[6] Maroudas A,Palla G,Gilav E.Racemization of aspartic acid in human articular cartilage[J].Connect Tissue Res,1992,28(3):161-169.
[7] Petersen E,Wågberg F,Angquist KA.Serum concentrations of elastin-derived peptides in patients with specific manifestations of atherosclerotic disease[J].Eur J Vasc Endovasc Surg,2002,24(5):440-444.
[8] Moore-Jones D,Perry HM Jr.Radioautographic localization of hydralazine-1-C-14 in arterial walls[J].Proc Soc Exp Biol Med,1966,122(2):576-579.
[9] Ohta K,Yamaguchi JI,Akimoto M,Fukushima K,Suwa T,Awazu S.Retention mechanism of imidazoles in connective tissue.Ⅰ.Binding to elastin[J].Drug Metab Dispos,1996,24(12):1291-1297.
[10] Ohta K,Akimoto M,Kohno Y,Fukushima K,Suwa T,Awazu S.Retention mechanism of imidazoles in connective tissue.Ⅱ.Activation of imidazoles in cupro-ascorbate system for irreversible binding formation with aortic tissuein vitro[J].Biol Pharm Bull,1998,21(3):308-310.
[11] Ohta K,Fukasawa Y,Yamaguchi J,Akimoto M,Kohno Y,Fukushima K,et al.Retention mechanism of imidazoles in connective tissue.Ⅲ.aldehyde adduct formation of a 4(5H)〔or 5(4H)〕-imidazolone productin vitro[J].Biol Pharm Bull,1998,21(9):958-963.
[12] Ohta K,Fukasawa Y,Yamaguchi J,Kohno Y,Fukushima K,Suwa T,et al.Retention mechanism of imidazoles in connective tissue.Ⅳ.Identification of a nucleophilic imidazolone metabolite in rats[J].Biol Pharm Bull,1998,21(12):1334-1337.
[13] Bombardier C,Laine L,Reicin A,Shapiro D,Burgos-Vargas R,Davis B,et al.Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis.VIGOR Study Group[J].N Engl J Med,2000,343(21):1520-1528.
[14] Food and Drug Administration.VIOXX®(rofecoxib tablets and oral suspension)[EB/OL].(2016-05-10)[2017-12-04].https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021042s033,021052s024lbl.pdf
[15] Food and Drug Administration.New Pediatric Labeling Information Database-Detai[lEB/OL].(2017-11-27)[2017-12-04]https://www.accessdata.fda.gov/scripts/sda/sdDetailNavigation.cfm?sd=labelingdatabase&id=14CE032258461B7DE053564DA8C071F2&rownum=512
[16] Doyle K,Lönn H,Käck H,Van de Poël A,Swallow S,Gardiner P,et al.Discovery of second generation reversible covalent DPP1 inhibitors leading to an oxazepane amidoacetonitrile based clinical candidate(AZD7986)[J].J Med Chem,2016,59(20):9457-9472.
