药源性线粒体毒性研究进展
2017-05-10凌嘉伟丁嘉欣姜昊玮江振洲张陆勇
凌嘉伟,丁嘉欣,陈 曦,姜昊玮,江振洲,王 涛,张陆勇,2
(1.中国药科大学江苏省新药筛选重点实验室,江苏南京 210009;2.广东药科大学药学院新药筛选与药效学评价中心,广东广州 510006)
线粒体在细胞能量代谢、凋亡途径、脂肪酸β-氧化、应激反应和稳态调节中发挥作用。自2000年以来,西立伐他汀(cerivastatin)和曲格列酮(troglitazone)的陆续撤市,使得药源性线粒体毒性的安全性评价得到重视。近年来,“线粒体毒性”和“线粒体功能障碍”这2个术语越来越多地出现在药物毒性或药物副作用的相关研究中,尤其在治疗人类免疫缺陷病毒感染和药源性肝损伤的研究中报道比较多。随着研究的发展,线粒体毒性机制逐渐被阐明。
1 线粒体结构与功能
线粒体是在所有真核细胞(红细胞除外)中发现的双层膜结构细胞器,它们的体积约占细胞体积的10%。线粒体是多型性细胞器,其结构变异取决于细胞类型、细胞周期阶段和细胞内代谢状态[1]。形态上,线粒体有2层不同的膜。外膜具有平滑边界,内膜形成内陷和遍布整个内腔的管状构造,被称为“嵴”。内膜和层状结构通过称为“嵴连接”的细管状连接相连。值得注意的是,内膜和外膜蛋白的含量和功能完全不同。
线粒体参与生物体内广泛的生物化学途径,包括氧化磷酸化(oxidative phosphorylation,OXPHOS)和糖、氨基酸、脂质以及内固醇代谢的关键步骤。线粒体通过OXPHOS产生ATP,提供细胞新陈代谢和钙离子缓冲所需的能量,并且是细胞内信号转导途径的重要“传感器”[2],可诱导细胞凋亡。线粒体具有2种不同的生物作用,一方面维持生物体生命力和活力,另一方面在调控细胞凋亡方面起着核心作用[3]。
2 线粒体损伤机制
线粒体毒性是许多疾病和毒性作用的潜在机制。一般来说,药物会在不同程度上损害线粒体功能。第一,药物可能直接影响线粒体DNA(mitochondrial DNA,mtDNA)并可能引起 mtDNA 突变[4]。第二,药物可能通过阻断或降低单个或多个呼吸链复合物的功能来影响整体呼吸链发挥作用[5]。通常,这将会导致能量产出降低,比如细胞内ATP的产生减少。第三,药物可能会异常增加线粒体活性氧(reactive oxygen species,ROS)的产生,从而增加氧化应激[6],也可以通过降低细胞器的抗氧化能力而间接增加氧化应激。氧化应激可增加自发性mtDNA突变的可能性,损害呼吸链功能,以及增加基质和线粒体膜间隙内的脂质或蛋白质的氧化。第四,这些药物的毒性可能降低线粒体内膜的膜电位,从而影响许多功能的正常发挥,甚至可能导致细胞器或细胞死亡[7]。第五,用于治疗心脏疾病的药物可能通过诱导多种凋亡途径诱导细胞凋亡,线粒体通透性转换孔(mitochondrial permeability transition pore,mPTP)开放是其启动机制之一[8]。除此之外,线粒体可能还存在其他损伤机制,但尚未阐明。
3 药源性线粒体毒性
长期以来,在设计适于实现所需生理效应的药物制剂中,对线粒体的生物能量学特征进行了较多的研究,例如,通过OXPHOS的解偶联来减轻体质量。然而,最近研究表明,线粒体是其他药物作用的非预期靶点,并且与大量药物的剂量限制性毒性有关。运用线粒体分子生物学和生物能量学理论可以解释药物诱导的线粒体毒性作用。这些非预期的毒性作用通常发生在线粒体功能的各个环节,比如抑制线粒体呼吸链酶活性、诱导线粒体氧化应激和OXPHOS解偶联作用等。
3.1 镇痛药
非甾体抗炎药(non-steroidal anti-inflammatory drugs,NSAID)常用于治疗各种疼痛。许多NSAID可能影响线粒体功能[9]。因为NSAID可导致线粒体功能障碍,其抑制环氧化酶而引起的花生四烯酸积累可能不利于心脏功能恢复[10]。吲哚美辛(indometacin)、双氯芬酸(diclofenac)和其他NSAID(包括选择性环氧化酶2抑制剂)在离体大鼠心脏中可引起呼吸作用解偶联,并减少ATP产生[9]。此外,在大鼠和新生大鼠心肌细胞中,NSAID可能通过抑制线粒体电子传递链而增加ROS的产生[11],对长期用药患者可能产生心脏毒性。
肝毒性是NSAID导致的一类罕见但重要的副作用。大多数NSAID中的二苯胺,在大鼠肝中可导致OXPHOS解偶联,降低肝ATP含量,引起线粒体肿胀从而诱导肝细胞损伤[12]。选择性环氧化酶2抑制剂引起的肝损伤,可能是其对ATP合成的直接抑制作用所致,在体外研究中,罗美昔布(lumiracoxib)的作用最强,其次是塞来昔布(celecoxib)和依托昔布(etoricoxib)[13]。双氯芬酸可引起特异性肝毒性。药物及其活性代谢物可与细胞蛋白质共价结合,引发特异反应[14]。在原代培养肝细胞的研究中,双氯芬酸及其代谢物能够抑制线粒体ATP的合成[5]。由于对乙酰氨基酚和阿司匹林不含二苯胺结构,二者可用于NSAID诱导肝毒性患者的治疗[15]。然而,体外研究表明,阿司匹林的主要代谢产物水杨酸盐增加了mPTP开放的敏感性。线粒体功能障碍也是对乙酰氨基酚诱导的肝毒性的重要决定因素。对乙酰氨基酚代谢物与线粒体蛋白质结合并诱导氧化应激,导致mPTP开放。经对乙酰氨基酚毒性剂量处理的人肝细胞表现出ROS产生增加[16]。这可能与肝细胞中参与线粒体呼吸链的相关基因表达发生改变有关。此外,线粒体抗氧化剂超氧化物歧化酶的表达降低,可能进一步导致ROS产生增加,引起进行性氧化应激[17]。由于肝毒性,尼美舒利(nimesulide)已经在一些国家撤市。研究表明,在离体培养的大鼠肝细胞和人肝癌细胞中,尼美舒利通过线粒体呼吸作用解偶联和诱导mPTP开放而影响线粒体ATP的产生,表明该药对线粒体有直接毒性作用[18]。
3.2 β受体阻滞剂
β受体阻滞剂是一类用于治疗心血管疾病的重要药物。已有研究表明,在有氧运动后,β2肾上腺素能受体可上调小鼠骨骼肌中线粒体生物合成调节因子过氧化物酶体增殖物激活受体γ辅激活因子1α(peroxisome proliferator-activated receptor-γ coactivator-1α,PGC-1α)的表达[19]。因此,使用非选择性β受体阻滞剂可能会削弱患者运动后骨骼肌线粒体的适应性和最大有氧呼吸能力[20]。然而,在离体大鼠骨骼肌线粒体的研究中,选择性β1阻滞剂美托洛尔(metoprolol)和阿替洛尔(atenolol)未发生上述变化。其中,普萘洛尔(propranolol)的脂质溶解度可能是其原因之一[21]。因此,建议心血管疾病患者进行锻炼时要避免使用非选择性β受体阻滞剂,否则可能起反作用[22]。
3.3 抗逆转录病毒药物
核苷类反转录酶抑制剂(nucleoside reverse transcriptase inhibitor,NRTI)的副作用中,乳酸中毒、高乳酸血症和脂肪代谢障碍综合征是由线粒体毒性造成的[23]。尽管结构差异导致不同程度的线粒体功能障碍,但是NRTI通常导致mtDNA缺失,导致线粒体呼吸受损,引起细胞内脂质积累和高乳酸血症[24]。依法韦仑(efavirenz,EFV)是一种重要的非核苷逆转录酶抑制剂,对线粒体呼吸链复合物Ⅰ的生物合成具有毒性作用,可以诱导人肝细胞中氧化应激的发生并显著增加线粒体质量。由于标准的抗逆转录病毒治疗方案是由EFV与2种NRTI联用组成,因此这些药物总体产生的毒性作用可能导致肝毒性[25]。EFV的神经精神不良反应也可能是由于神经元中线粒体改变和ATP产生减少所致[26]。对人类免疫缺陷病毒感染患者使用NRTI治疗,也可能加速骨骼肌中细胞mtDNA突变而导致早衰[27]。
3.4 抗癌药物
多柔比星是一种蒽环类抗肿瘤抗生素,用于治疗多种血液恶性肿瘤和实体瘤。导致心肌病是该药物使用受限的一个重要原因。诱导线粒体功能障碍是多柔比星心脏毒性的主要原因[28]。多柔比星引起心脏线粒体功能障碍的原因可能是由于ROS产生增加[29],增加一氧化氮的产生[30],并且增加铁离子在线粒体的积聚所致[28]。多柔比星诱导的线粒体功能障碍也可能是长期儿童癌症患者并发骨骼肌功能障碍的基础。用多柔比星和地塞米松处理4个周期的非荷瘤小鼠模型显示,骨骼肌量和线粒体呼吸减少,并且ROS产生增加。表明在骨骼肌功能障碍中,可能存在线粒体能量合成障碍[31]。多柔比星还通过促进血小板凋亡引起血小板减少症,这可能是由于线粒体ROS剂量依赖性增加所致[32]。血小板线粒体ROS水平升高的机制尚不清楚。
顺铂是另一种常见的可用于实体肿瘤的抗癌药物,可引起剂量依赖性的肾毒性。顺铂诱导的肾毒性涉及ROS的形成,ATP的减少,抵御抗氧化剂作用和线粒体呼吸[33]。ROS生成增加可能是由于顺铂对线粒体呼吸链复合物Ⅰ的抑制作用[34]。在顺铂诱导的肾损伤小鼠模型中,顺铂改变了肾线粒体结构和功能。线粒体功能受损可能是由于线粒体质量下降和酶活性降低所致[35]。有报道表明,带正电荷的顺铂代谢物优先积累在带负电的线粒体内。因此,线粒体富集的肾近端肾小管细胞受到的影响最大[36]。
3.5 降血脂药物
他汀类药物是最常用的降血脂药。肝毒性和肌病虽然不常见,确是他汀类药物的严重副作用。线粒体是肌病发病机制中的重要靶点。已有研究报道,他汀类药物对心脏和骨骼肌的作用相反。他汀类药物通过“线粒体毒性兴奋机制”,即通过刺激线粒体生物发生调节剂PGC-1α和β,以及提高抗氧化能力来抵抗心脏中的轻度氧化应激,从而保护心肌。但由于骨骼肌中抵抗他汀类药物诱导高氧化应激的抗氧化能力较弱,他汀类药物可诱导骨骼肌线粒体功能障碍、线粒体生物发生受损和肌病。氧化还原电位的改变可能是他汀类药物毒性作用的原因[37]。研究报道,与健康对照组相比,服用辛伐他汀(simvastatin)的高胆固醇血症患者肌肉活组织葡萄糖耐量受损,最大OXPHOS能力受损,辅酶Q10蛋白含量下降[38]。
对他汀类药物诱发肌病患者的骨骼肌活检的回顾性研究显示,他汀类药物治疗与骨骼肌mtDNA的消耗有关[39]。从健康志愿者的肌肉活组织检查中获得的骨骼肌细胞的研究表明,辛伐他汀和洛伐他汀(lovastatin)诱导线粒体凋亡途径存在浓度和时间依赖性。这种作用主要是通过激活钙蛋白酶、胱天蛋白酶3和胱天蛋白酶9[40],增加细胞内钙离子浓度来介导的。除他汀类药物外,贝特类药物,特别是与他汀类药物联用后会引起肌病[41]。贝特类药物联合他汀类药物可能会加重横纹肌溶解症[42]。研究表明,非诺贝特(fenofibrate)和氯贝丁酯(clofibrate,另一类主要用于高甘油三酯血症的降脂药物)可明显抑制线粒体呼吸链复合物Ⅰ的活性[43]。
3.6 抗癫痫药
许多抗癫痫药物都能不同程度地引起线粒体功能障碍。其中,丙戊酸(valproic acid)是一种广谱抗癫痫药,可引起线粒体毒性[44],肝毒性是其最显著的副作用[45]。对线粒体疾病患者的研究表明,丙戊酸诱导的肝毒性与mtDNA聚合酶γ突变存在相关性[46]。研究提出假设,丙戊酸的主要代谢物丙戊酸辅酶A的积累抑制琥珀酰辅酶A连接酶,继而损害核苷二磷酸激酶的活性,引起线粒体中的核苷酸不平衡[47],导致mtDNA的耗竭。用丙戊酸处理大鼠肝线粒体和HepG2细胞后发现,药物通过抑制呼吸链复合物Ⅱ,诱导脂质过氧化和线粒体膜电位受损导致细胞色素c的释放增加,引起ROS产生增加,进而激活细胞死亡途径[44]。除丙戊酸钠外,苯妥英(phenytoin)、卡马西平和苯巴比妥等芳香族抗癫痫药物也可诱导线粒体功能障碍。研究报道,它们与线粒体功能障碍有关的体外活性:苯妥英>苯巴比妥>卡马西平[48]。与卡马西平相比,奥卡西平(oxcarbazepine)能够增加ROS产生,减少ATP合成并且降低线粒体膜电位,其对神经元的毒性相对更高[49]。此外,体外研究还显示,唑尼沙胺(zonisamide)和托吡酯(topiramate)抑制线粒体锌酶和人碳酸酐酶[50]。这些线粒体效应在临床病例上的相关性尚不明确。
3.7 抗精神病药
典型和非典型抗精神病药物都会对线粒体功能产生影响[51]。与典型药物相比,非典型抗精神病药物的锥体外系副作用(如迟发性运动障碍)的风险较低。体外研究数据表明,抗精神病药物会影响线粒体功能,特别是抑制线粒体呼吸链复合物Ⅰ活性,并且这与其产生锥体外系副作用(包括迟发性运动障碍)有关[52]。利用人脑微血管内皮细胞的研究证实了精神安定药(典型抗精神病药)的这些作用,其中复合物Ⅰ和Ⅲ的活性能被所有精神安定药抑制[51]。此外,吩噻嗪(phenthiazine)衍生物还通过mPTP开放和细胞色素c释放诱导细胞死亡[53-54]。氯氮平(cozapine)是一种非典型的抗精神病药物,可引起代谢综合征。通过对胰岛素应答和肥胖相关的培养细胞的研究发现,其中涉及的机制包括线粒体形态改变、线粒体膜电位降低和ATP耗竭。氯氮平还增加了促炎症细胞因子的产生[55]。这对糖尿病和心血管疾病患者来说尤其需要重视。
3.8 抗抑郁药
许多体内外研究表明,频繁使用抗抑郁药导致线粒体功能抑制和氧化应激增加[56]。辅酶Q10缺陷是阿米替林(amitriptyline)诱导的线粒体毒性的标志。阿米替林诱导的线粒体毒性也包括线粒体蛋白表达减少、线粒体含量降低、ATP水平下降、ROS产生增加和mPTP开放[56-57]。尽管ROS产生的增加可能是由于辅酶Q水平的降低,但是具体的分子机制还需要进一步研究。由于这些功能失调的特征也可能参与抑郁症的病理生理学,所以阿米替林的治疗可能会使得病情恶化[57]。其他抗抑郁药如氯丙咪嗪(clomipraminum)、地昔帕明(desipramine)、噻奈普丁(tianeptine)和氟西汀(fluoxetine),同样对线粒体产生作用[56,58-59]。氟西汀和噻奈普汀具有抑制线粒体呼吸的作用,而氯丙咪嗪、地昔帕明和诺氟西汀(norfluoxetine)则通过降低线粒体膜电位和抑制线粒体复合物活性而显示凋亡效应[58-59]。这些作用是否与其治疗作用或副作用有关并不清楚。研究表明,舍曲林(sertraline)和曲唑酮(trazodone)对线粒体的作用与严重肝毒性副作用有关[60-61]。舍曲林导致ATP耗竭,诱导mPTP开放和抑制线粒体呼吸链[60],在长期给药后会导致不可逆的线粒体损伤和细胞死亡。由于药物引起的肝毒性,另一种抗抑郁药萘发扎酮(nefazadone)被撤市。在体外人肝细胞中,萘发扎酮能够抑制线粒体膜电位和线粒体呼吸链复合物Ⅰ和Ⅳ活性[62]。
4 结语
综上所述,很多药物都可以直接或间接地对线粒体产生毒性作用(表1)。一些仅在体外研究中发现,另一些则是在临床患者中观察到。药物诱导的线粒体功能障碍往往导致一些药物的副作用。因此,药物线粒体毒性分析筛选成为药物临床前研究的重要一环,并且是上市后需要实时关注的指标。同时,在传统的体内实验模型的基础上,需要开发更多种属的动物模型或细胞系,选取合适的线粒体毒性靶标,应用高通量高内涵技术,进行高效率的大规模线粒体毒性分析。通过对大量化合物的线粒体毒性筛选鉴定,得出毒性-结构关系,为未知化合物的线粒体毒性提供预测。
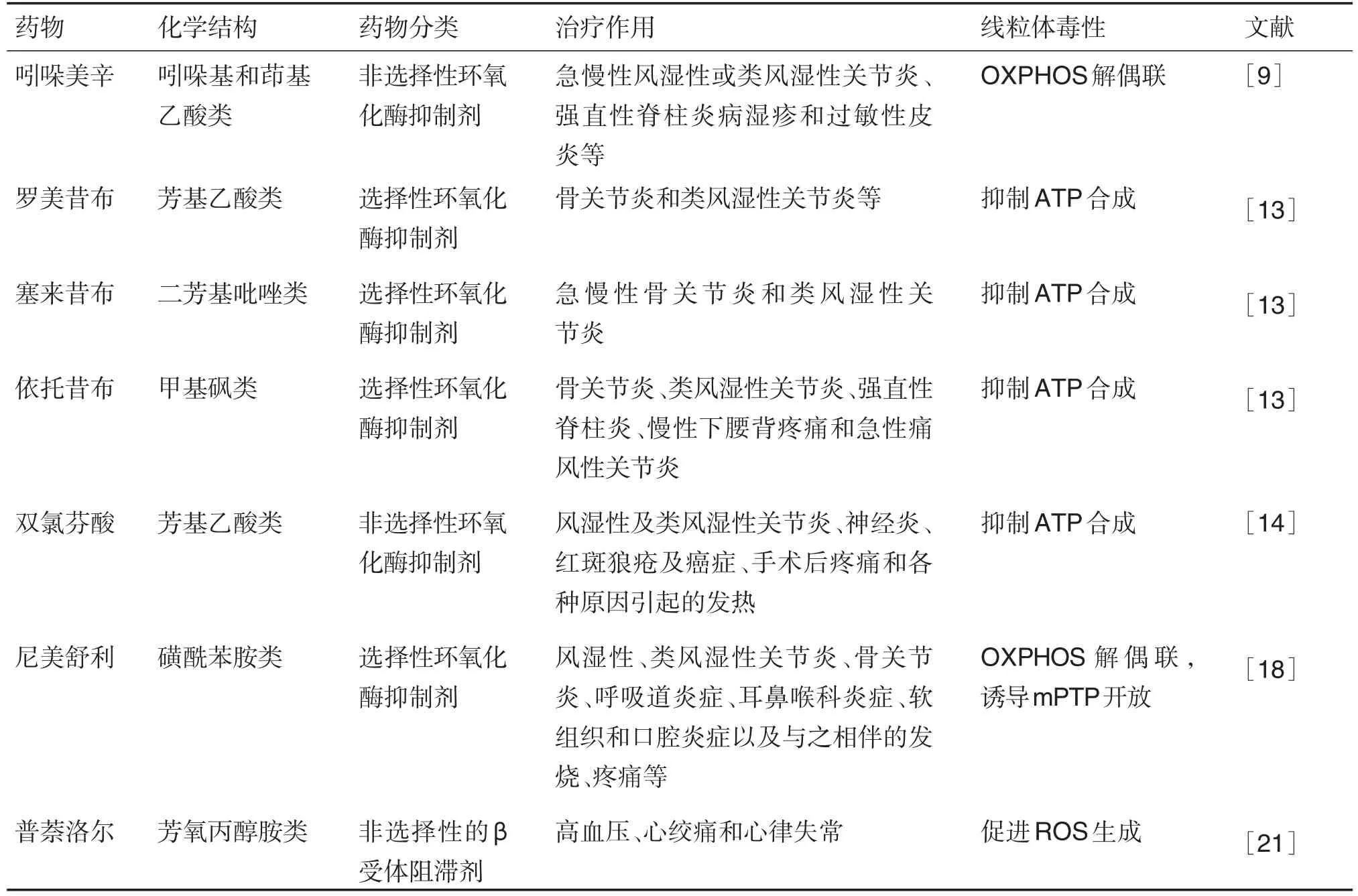
表1 引起线粒体毒性的药物及其机制.
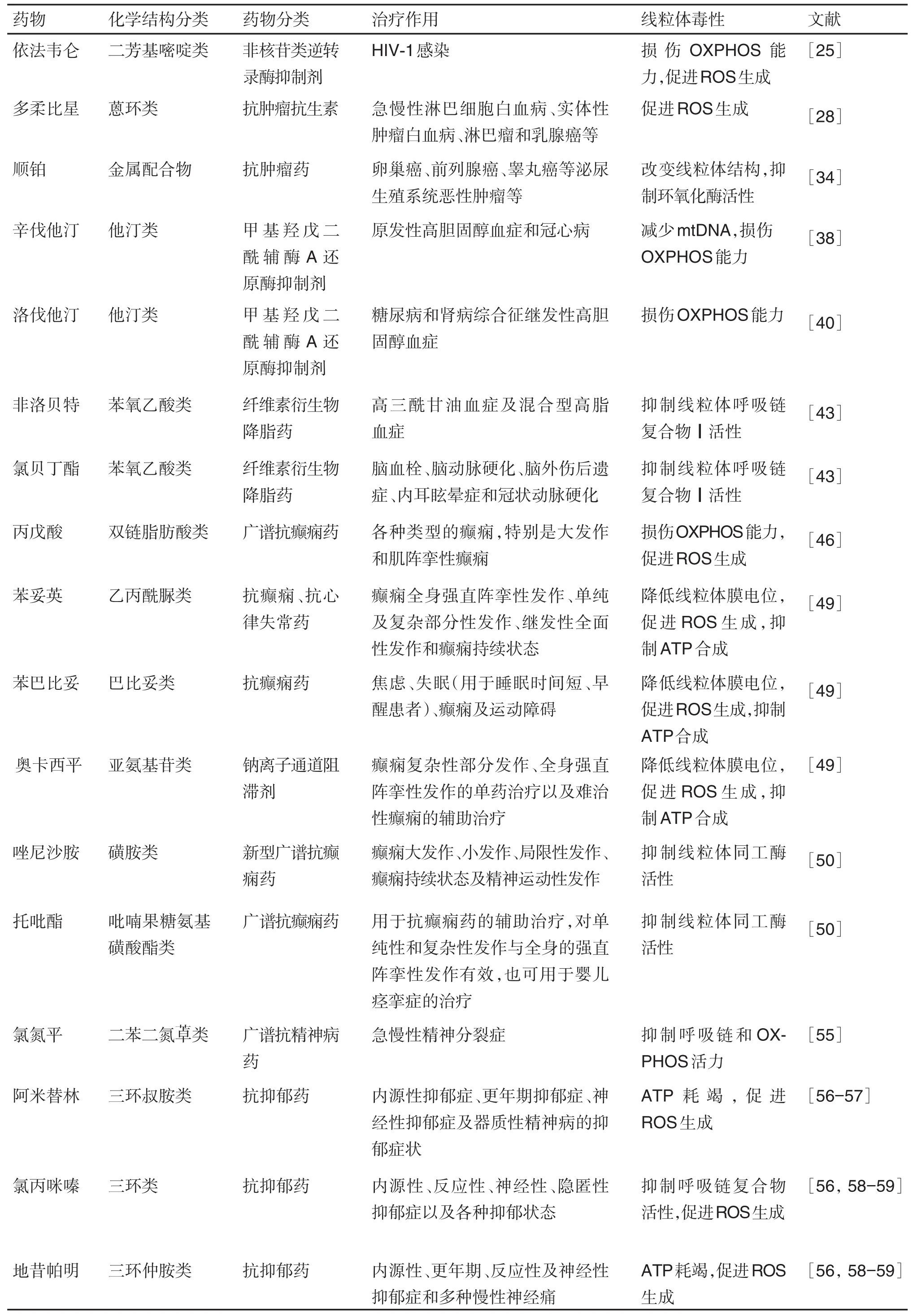
续表1
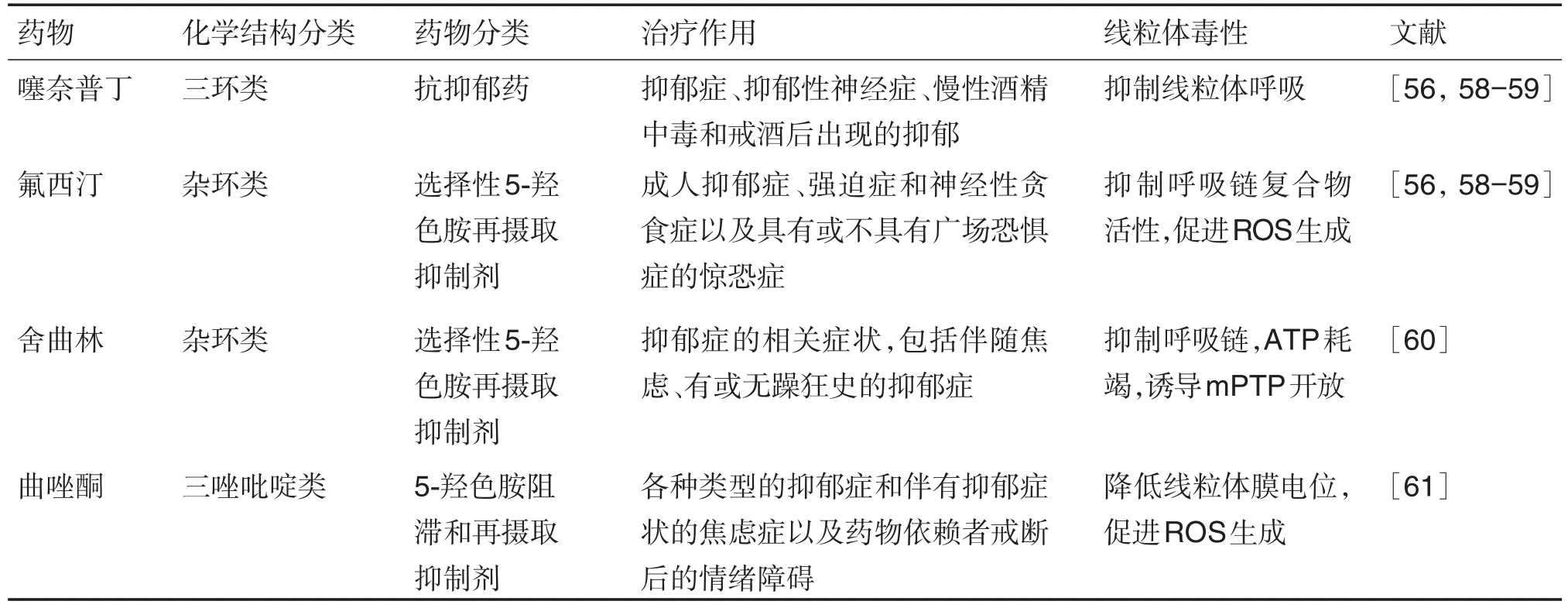
续表1
参考文献:
[1] Srivastava N,Pande M.Mitochondrion:features,functions and comparative analysis of specific probes in detecting sperm cell damages [J].Asian Pac J Reprod,2016,5(6):421-429.
[2] Zolkipli-Cunningham Z,Falk MJ.Clinical effects of chemical exposures on mitochondrial function[J].Toxicology,2017,391:90-99.
[3] Kroemer G,Galluzzi L,Brenner C.Mitochondrial membrane permeabilization in celldeath [J].Physiol Rev,2007,87(1):99-163.
[4] Alexeyev M,Shokolenko I,Wilson G,Ledoux S.The maintenance of mitochondrial DNA integritycritical analysis and update[J/OL].CSH Perspect Biol,2013,5(5):a012641(2013-05).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3632056/
[5] Syed M,Skonberg C,Hansen SH.Mitochondrial toxicity of diclofenac and its metabolites via inhibition of oxidative phosphorylation(ATP synthesis)in rat liver mitochondria:possible role in drug induced liver injury(DILI)[J].Toxicol In Vitro,2016,31:93-102.
[6] Fu PP,Xia Q,Hwang HM,Ray PC,Yu H.Mechanisms of nanotoxicity:generation of reactive oxygen species[J].J Food Drug Anal,2014,22(1):64-75.
[7] Martínez-Reyes I,Diebold LP,Kong H,Schieber M,Huang H,Hensley CT,et al.TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions[J].Mol Cell,2016,61(2):199-209.
[8] Roqanian S, Meratan AA, Ahmadian S,Shafizadeh M,Ghasemi A,Karami L.Polyphenols protect mitochondrial membrane against permeabilization induced by HEWL oligomers:possible mechanism of action[J].Int J Biol Macromol,2017,103:709-720.
[9] Moreno-Sánchez R,Bravo C,Vásquez C,Ayala G,Silveira LH,Martínez-Lavín M.Inhibition and uncoupling of oxidative phosphorylation by nonsteroidal anti-inflammatory drugs:study in mitochondria,submitochondrial particles,cells,and whole heart[J].Biochem Pharmacol,1999,57(7):743-752.
[10] Fosslien E.Cardiovascular complications of nonsteroidal anti-inflammatory drugs[J].Ann Clin Lab Sci,2005,35(4):347-385.
[11] Ghosh R,Hwang SM,Cui Z,Gilda JE,Gomes AV.Different effects of the nonsteroidal anti-inflammatory drugs meclofenamate sodium and naproxen sodium on proteasome activity in cardiac cells[J].J Mol Cell Cardiol,2016,94:131-144.
[12] Ghosh R,Goswami SK,Feitoza LF,Hammock B,Gomes AV.Diclofenac induces proteasome and mitochondrial dysfunction in murine cardiomyocytes and hearts[J].Int J Cardiol,2016,223:923-935.
[13] Syed M,Skonberg C,Hansen SH.Mitochondrial toxicity of selective COX-2 inhibitors via inhibition of oxidative phosphorylation(ATP synthesis)in rat liver mitochondria[J].Toxicol In Vitro,2016,32:26-40.
[14] Ghosh R,Alajbegovic A,Gomes AV.NSAIDs and cardiovascular diseases:role of reactive oxygen species[J/OL].Oxid Med Cell Longev,2015,2015:536962(2015-09-20).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4592725/
[15] Soleimanpour M,Imani F,Safari S,Sanaie S,Soleimanpour H,Ameli H,et al.The role of non-steroidal anti-inflammatory drugs(NSAIDs)in the treatment of patients with hepatic disease:a review article[J/OL].Anesth Pain Med,2016,6(4):e37822(2016-08-10).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5100664/
[16] González LT,Minsky NW,Espinosa LE,Aranda RS,Meseguer JP,Pérez PC.In vitroassessment of hepatoprotective agents against damage induced by acetaminophen and CCl4[J/OL].BMC Comple⁃ment Altern Med,2017,17(1):39(2017-01-13).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5234107/
[17] Jiang J,Briedé JJ,Jennen DG,Van Summeren A,Saritas-Brauers K,Schaart G,et al.Increased mitochondrial ROS formation by acetaminophen in human hepatic cells is associated with gene expression changes suggesting disruption of the mitochondrial electron transport chain[J].Toxicol Lett,2015,234(2):139-150.
[18] Jaeschke H, McGill MR, Ramachandran A.Oxidantstress, mitochondria, andcelldeath mechanisms in drug-induced liver injury:lessons learned from acetaminophen hepatotoxicity[J].Drug Metab Rev,2012,44(1):88-106.
[19] Brandt N,Dethlefsen MM,Bangsbo J,Pilegaard H.PGC-1α and exercise intensity dependent adaptations in mouse skeletal muscle[J/OL].PLoS One,2017,12(10):e0185993(2017-10-19).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5648136/
[20] Robinson MM,Bell C,Peelor FF 3rd,Miller BF.β-adrenergic receptor blockade blunts postexercise skeletal muscle mitochondrial protein synthesis rates in humans[J].Am J Physiol Regul Integr Comp Physiol,2011,301(2):R327-R334.
[21] Dreisbach AW,Greif RL,Lorenzo BJ,Reidenberg MM.Lipophilic beta-blockers inhibit rat skeletal muscle mitochondrial respiration[J].Pharmacology,1993,47(5):295-299.
[22] Bacurau AV,Cunha TF,Souza RW,Voltarelli VA,Gabriel-Costa D,Brum PC.Aerobic exercise and pharmacological therapies for skeletal myopathy in heart failure:similarities and differences[J/OL].Oxid Med Cell Longev,2016,2016:4374671(2016-01-19).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4745416/
[23] Foli Y,Ghebremichael M,Li M,Paintsil E.Upregulation of apoptosis pathway genes in peripheral blood mononuclear cells of HIV-infected individuals with antiretroviral therapy-associated mitochondrial toxicity [J/OL].Antimicrob Agents Chemother,2017,61(8):e00522-17(2017-07-25).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5527626/
[24] Tang YW,Ou CY.Past,present and future molecular diagnosis and characterization of human immunodeficiency virus infections[J/OL].Emerg Microbes Infect,2012,1(8):e19(2012-08-22).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3630918/
[25] Apostolova N, Gomez-Sucerquia LJ,Moran A,Alvarez A,Blas-Garcia A,Esplugues JV.Enhanced oxidative stress and increased mitochondrial mass during efavirenz-induced apoptosis in human hepatic cells[J].Br J Pharmacol,2010,160(8):2069-2084.
[26] Funes HA,Blas-Garcia A,Esplugues JV,Apostolova N.Efavirenz alters mitochondrial respiratory function in cultured neuron and glial cell lines[J].J Antimi⁃crob Chemother,2015,70(8):2249-2254.
[27] Payne BA,Wilson IJ,Hateley CA,Horvath R,Santibanez-Koref M,Samuels DC,et al.Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations[J].Nat Genet,2011,43(8):806-810.
[28] Ichikawa Y,Ghanefar M,Bayeva M,Wu R,Khechaduri A,Naga Prasad SV,et al.Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation[J].J Clin Invest,2014,124(2):617-630.
[29] Kavazis AN,Morton AB,Hall SE,Smuder AJ.Effects of doxorubicin on cardiac muscle subsarcolemmal and intermyofibrillar mitochondria[J].Mito⁃chondrion,2017,34:9-19.
[30] Fogli S,Nieri P,Breschi MC.The role of nitric oxide in anthracycline toxicity and prospects for pharmacologic prevention of cardiac damage[J].FASEB J,2004,18(6):664-675.
[31] Gouspillou G,Scheede-Bergdahl C,Spendiff S,Vuda M,Meehan B,Mlynarski H,et al.Anthracycline-containing chemotherapy causes long-term impairment of mitochondrial respiration and increased reactive oxygen species release in skeletal muscle[J/OL].Sci Rep,2015,5:8717(2015-03-03).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4346812/
[32] Wang Z,Wang J,Xie R,Liu R,Lu Y.Mitochondria-derived reactive oxygen species play an important role in doxorubicin-induced platelet apoptosis[J].Int J Mol Sci,2015,16(5):11087-11100.
[33] Peres LA,da Cunha AD Jr.Acute nephrotoxicity of cisplatin:molecular mechanisms[J].J Bras Nefrol,2013,35(4):332-340.
[34] Kruidering M, Van de Water B, de Heer E,Mulder GJ, Nagelkerke JF. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells:mitochondrial dysfunction by inhibition of complexesⅠto Ⅳ of the respiratory chain[J].J Pharmacol Exp Ther,1997,280(2):638-649.
[35] Zsengellér ZK,Ellezian L,Brown D,Horváth B,Mukhopadhyay P,Kalyanaraman B,et al.Cisplatin nephrotoxicity involves mitochondrial injury with impaired tubular mitochondrial enzyme activity[J].J Histochem Cytochem,2012,60(7):521-529.
[36] Hong JY,Hara K,Kim JW,Sato EF,Shim EB,Cho KH.Minimal systems analysis of mitochondriadependent apoptosis induced by cisplatin[J].Korean J Physiol Pharmacol,2016,20(4):367-378.
[37] Bouitbir J,Charles AL,Echaniz-Laguna A,Kindo M,Daussin F,Auwerx J,et al.Opposite effects of statins on mitochondria of cardiac and skeletal muscles:a‘mitohormesis’mechanism involving reactive oxygen species and PGC-1[J].Eur Heart J,2012,33(11):1397-1407.
[38] Ramachandran R,Wierzbicki AS.Statins,muscle disease and mitochondria[J/OL].J Clin Med,2017,6(8):75(2017-07-25).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5575577/
[39] Stringer HA, Sohi GK,Maguire JA,Côté HC.Decreased skeletal muscle mitochondrial DNA in patients with statin-induced myopathy[J].J Neurol Sci,2013,325(1-2):142-147.
[40] Abdulrazaq M,Hamdan F,Al-Tameemi W.Electrophysiologic and clinico-pathologic characteristics of statin-induced muscle injury[J].Iran J Basic Med Sci,2015,18(8):737-744.
[41] Hodel C.Myopathy and rhabdomyolysis with lipidlowering drugs[J].Toxicol Lett,2002,128(1-3):159-168.
[42] Shek A,Ferrill MJ.Statin-fibrate combination therapy[J].Ann Pharmacother,2001,35(7-8):908-917.
[43] Brunmair B,Lest A,Staniek K,Gras F,Scharf N,Roden M,et al.Fenofibrate impairs rat mitochondrial function by inhibition of respiratory complexⅠ[J].J Pharmacol Exp Ther,2004,311(1):109-114.
[44] Komulainen T,Lodge T,Hinttala R,Bolszak M,Pietilä M,Koivunen P,et al.Sodium valproate induces mitochondrial respiration dysfunction in HepG2in vitrocell model[J].Toxicology,2015,331:47-56.
[45] Jafarian I,Eskandari MR,Mashayekhi V,Ahadpour M,Hosseini MJ.Toxicity of valproic acid in isolated rat liver mitochondria[J].Toxicol Mech Methods,2013,23(8):617-623.
[46] Kudin AP,Mawasi H,Eisenkraft A,Elger CE,Bialer M,Kunz WS.Mitochondrial liver toxicity of valproic acid and its acid derivatives is related to inhibition of α-lipoamide dehydrogenase[J/OL].Int J Mol Sci,2017,18(9):1912(2017-09-06).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5618561/
[47] Luís PB,Ruiter J,IJlst L,de Almeida IT,Duran M,Wanders RJ,et al.Valproyl-CoA inhibits the activity of ATP-and GTP-dependent succinate:CoA ligases[J].J Inherit Metab Dis,2014,37(3):353-357.
[48] AhmadianE, BabaeiH, Mohajjel NayebiA,Eftekhari A,Eghbal MA.Venlafaxine-induced cytotoxicity towards isolated rat hepatocytes involves oxidative stress and mitochondrial/lysosomal dysfunction[J].Adv Pharm Bull,2016,6(4):521-530.
[49] Araújo IM,Ambrósio AF,Leal EC,Verdasca MJ,Malva JO,Soares-da-Silva P,et al.Neurotoxicity induced by antiepileptic drugs in cultured hippocampal neurons:a comparative study between carbamazepine, oxcarbazepine, andtwonew putative antiepileptic drugs,BIA 2-024 and BIA 2-093[J].Epilepsia,2004,45(12):1498-1505.
[50] De Simone G,Di Fiore A,Menchise V,Pedone C,Antel J,Casini A,et al.Carbonic anhydrase inhibitors.Zonisamide is an effective inhibitor of the cytosolic isozyme Ⅱ and mitochondrial isozymeⅤ:solution and X-ray crystallographic studies[J].Bioorg Med Chem Lett,2005,15(9):2315-2320.
[51] Elmorsy E,Smith PA.Bioenergetic disruption of human micro-vascular endothelial cells by antipsychotics [J].BiochemBiophys Res Commun,2015,460(3):857-862.
[52] Goh S,Dong Z,Zhang Y,DiMauro S,Peterson BS.Mitochondrial dysfunction as a neurobiological subtype of autism spectrum disorder:evidence from brain imaging[J].JAMA Psychiatry,2014,71(6):665-671.
[53] Cruz TS,Faria PA,Santana DP,Ferreira JC,Oliveira V,Nascimento OR,et al.On the mechanisms of phenothiazine-induced mitochondrial permeability transition:thiol oxidation,strict Ca2+dependence,and Cyt c release[J].Biochem Pharmacol,2010,80(8):1284-1295.
[54] de Faria PA,Bettanin F,Cunha RL,Paredes-Gamero EJ,Homem-de-Mello P,Nantes IL,et al.Cytotoxicity of phenothiazine derivatives associated with mitochondrial dysfunction:a structure-activity investigation[J].Toxicology,2015,330:44-54.
[55] Contreras-Shannon V,Heart DL,Paredes RM,Navaira E,Catano G,Maffi SK,et al.Clozapineinduced mitochondria alterations and inflammation in brain and insulin-responsive cells[J/OL].PLoS One,2013,8(3):e59012(2013-05-20).https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3604003/
[56] Lee MY,Hong S,Kim N,Shin KS,Kang SJ.Tricyclic antidepressants amitriptyline and desipramine induced neurotoxicity associated with Parkinson′s disease[J].Mol Cells,2015,38(8):734-740.
[57] Moreno-Fernández AM,Cordero MD,Garrido-Maraver J,Alcocer-Gómez E,Casas-Barquero N,Carmona-López MI,et al.Oral treatment with amitriptyline inducescoenzymeQ deficiency and oxidative stress in psychiatric patients[J].J Psychiatr Res,2012,46(3):341-345.
[58] Abdel-Razaq W,Kendall DA,Bates TE.The effects of antidepressants on mitochondrial function in a model cell system and isolated mitochondria[J].Neurochem Res,2011,36(2):327-338.
[59] Głombik K,Stachowicz A,Olszanecki R,S′lusarczyk J,Trojan E,Lasoń W,et al.The effect of chronic tianeptine administration on the brain mitochondria:direct links with an animal model of depression[J].Mol Neurobiol,2016,53(10):7351-7362.
[60] Li Y, Couch L, Higuchi M, Fang JL, Guo L.Mitochondrial dysfunction induced by sertraline,an antidepressant agent[J].Toxicol Sci,2012,127(2):582-591.
[61] Taziki S,Sattari MR,Eghbal MA.Mechanisms of trazodone-induced cytotoxicity and the protective effects of melatonin and/or taurine toward freshly isolated rat hepatocytes[J].J Biochem Mol Toxicol,2013,27(10):457-462.
[62] Dykens JA,Jamieson JD,Marroquin LD,Nadanaciva S,Xu JJ,Dunn MC,et al.In vitroassessment of mitochondrial dysfunction and cytotoxicity of nefazodone,trazodone,and buspirone[J].Toxicol Sci,2008,103(2):335-345.
