RNA-Seq高通量测序技术在果树功能基因组学研究的应用进展
2017-03-31黄丽娜张奋强姜梦嫣周义杰陈思源王有年杨明峰马兰青
刘 欢, 黄丽娜, 张奋强, 姜梦嫣, 周义杰, 陈思源, 王有年, 杨明峰*, 马兰青,3*
1.北京农学院植物科学技术学院, 农业部华北都市农业重点实验室, 北京 102206;2.北京农学院生物科学与工程学院, 农业部华北都市农业重点实验室, 北京 102206;3.北京农学院, 北京林果业生态环境功能提升协同创新中心, 北京 102206
RNA-Seq高通量测序技术在果树功能基因组学研究的应用进展
刘 欢1, 黄丽娜2, 张奋强1, 姜梦嫣2, 周义杰1, 陈思源2, 王有年2, 杨明峰2*, 马兰青2,3*
1.北京农学院植物科学技术学院, 农业部华北都市农业重点实验室, 北京 102206;2.北京农学院生物科学与工程学院, 农业部华北都市农业重点实验室, 北京 102206;3.北京农学院, 北京林果业生态环境功能提升协同创新中心, 北京 102206
RNA-Seq又称为“转录组测序技术”,它能够从整体水平研究物种基因功能以及基因结构,揭示特定生物学过程。应用该项技术能有效地解决因果树基因组庞大等问题,有效地揭示和阐明植物体内次生代谢生物合成途径及代谢机制。主要论述了RNA-Seq技术的应用方法与流程,通过对测序数据的处理分析与比对等方法,分析了RNA-Seq技术在果树新基因挖掘、次生代谢产物合成途径的应用进展,基于RNA-Seq的高通量测序技术对于发现和揭示具有重要生物学功能和经济价值的次生代谢产物合成关键基因及其调控机制提供了可能。
高通量测序;RNA-Seq;果树;次生代谢产物
基因测序技术也称DNA测序技术,是现代生物学研究中重要的手段之一。截至目前,基因测序技术一共经历了三个发展阶段。继1963年Sanger等人第一次完成胰岛素51个氨基酸序列的测定后,1975年Sanger和Maxam-Gilbert等又建立了核酸序列测定的方法——双脱氧末端终止法,将其称之为第一代测序技术。随后,哈佛大学遗传学家George Church和454 Life Sciences公司 Jonathan Rothberg掀起的“新一代测序技术(NGS)” 也被称为二、三代测序技术,均以高通量为共同特征。Roche公司的454测序平台、Illumina公司的Solexa测序系统以及ABI公司的SOLID测序系统标志着第二代测序技术诞生。Helicos公司的Heliscope单分子测序仪、Pacific Biosciences公司的SMRT技术、Oxford Nanopore Technologies公司研究的纳米孔单分子技术,被认为是第三代测序技术。RNA-Seq对研究不同环境下植物体内基因表达差异的技术研究方面具有重要作用[1]。该技术结合了构建转录组测序文库的实验方法与DGE (digital gene expression TagProfiling) 的信息分析手段,具有高通量的特点,开辟了功能基因组学研究的新纪元[2]。
转录组是连接基因调控和功能蛋白质组的纽带,RNA-Seq与传统的cDNA文库构建及EST测序相比[3],可在较短时间、较少人工和更低成本的情况下获得大量序列信息,尤其是可获得低表达丰度的基因序列,对研究植物抗逆性的基因表达方式有重要意义[4]。针对遗传背景复杂、杂合度高的果树研究瓶颈,RNA-Seq结合自身优势,已成为果树学研究的新突破点。据统计:甜橙[5,6]、苹果[7,8]、草莓[9]、猕猴桃[10,11]、蓝莓[12]、荔枝[13]、火龙果[14]、李子[15]、树莓[16]等果树已应用这一技术获得了高通量的转录本序列信息及表达量数据,并为其在基因功能验证及预测、生物体内代谢与调控、果实发育与品质形成、环境胁迫响应等方面的研究奠定了基础。本文先对RNA-Seq测序技术的研究情况进行总述,再结合转录组测序技术在果树功能基因组学中的应用研究进行分析,最后小结并展望今后研究中面临的问题与趋势,以期为相关研究提供参考。
1 RNA-Seq技术的应用与方法
高通量测序技术是对传统测序一次革命性的改变,一次性对几十万到几百万条DNA分子进行序列测定,高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能。在转录组水平上进行转录组测序(whole transcriptome resequencing),也称为RNA-Seq,是研究基因功能及结构研究的出发点;从而可以相继开展可变剪接、转录本变异研究(如基因融合编码区SNP研究)、非编码区域功能研究(non-coding RNA研究和microRNA前体研究等)[11]等方面的研究。
1.1 RNA-Seq的实验及分析方法
转录组测序的主要目标是获得某个生物体或某个组织在特定条件下(如经过高温、光照、水分、盐分胁迫、各种激素的诱导等)表达的全部基因序列,实验流程见图1[17]。

图1 RNA-Seq的实验流程Fig.1 Experiment pipeline of RNA-Seq.
1.1.1 测序文库的制备方法[17]提取植物样品总RNA,然后用带有Oligo (dT)的磁珠富集mRNA,向得到的mRNA中加入适量fragmentation buffer高温条件下使其片断化,再以片断后的mRNA为模板,合成cDNA,经过磁珠纯化、末端修复、在3’末端加A尾、测序接头与dsDNA连接并纯化,进行PCR扩增。用Agilent 2100 Bioanalyzer 和ABI StepOnePlus Real-time PCR System 进行质量和产量检测,文库质控合格后进行测序。
1.1.2 数据分析 由于所测数据结果庞大而冗杂,测序所得的原始数据(rawreads 或raw data) 首先进行质量筛选,以确定测序数据是否适用于后续分析[1]。经过滤得到的clean reads与参考序列做对比进行筛选,质量分布通常大于30(小于20为质量较低),并对筛选出的样品间差异表达基因进行GO功能注释,KEGG生物体内代谢分析,基因功能性富集分析(Q-value≤0.05)[18]。
通过RT-PCR、qRT-PCR验证潜在样品间差异表达上、下调基因的准确性,进一步阐述他们的作用机制。
1.1.3 RNA-Seq在果树上的应用 由于果树存在发育周期长、遗传背景复杂、基因组庞大等特点,相对草本植物与动物的生物合成途径研究相对滞后,主要是木本植物基因进化、遗传特性以及生态等因素使其基因组信息严重缺乏。转录组研究是生物合成途径、关键基因功能及结构研究的基础和出发点,通过转录组测序技术(RNA-Seq),能够全面且快速地获得某一物种特定组织或器官在某一状态下的几乎所有转录本序列信息。Zhang等[19]于1997 年最早提出了转录组的概念,这一概念的提出有效地解决了对遗传背景复杂、杂合度高木本植物型果树的研究瓶颈。转录组测序是用于研究对象在某一功能状态下所能转录出来的所有RNA的总和,主要包括 mRNA和非编码RNA(rRNA、tRNA、snRNA、snoRNA和microRNA 等)。
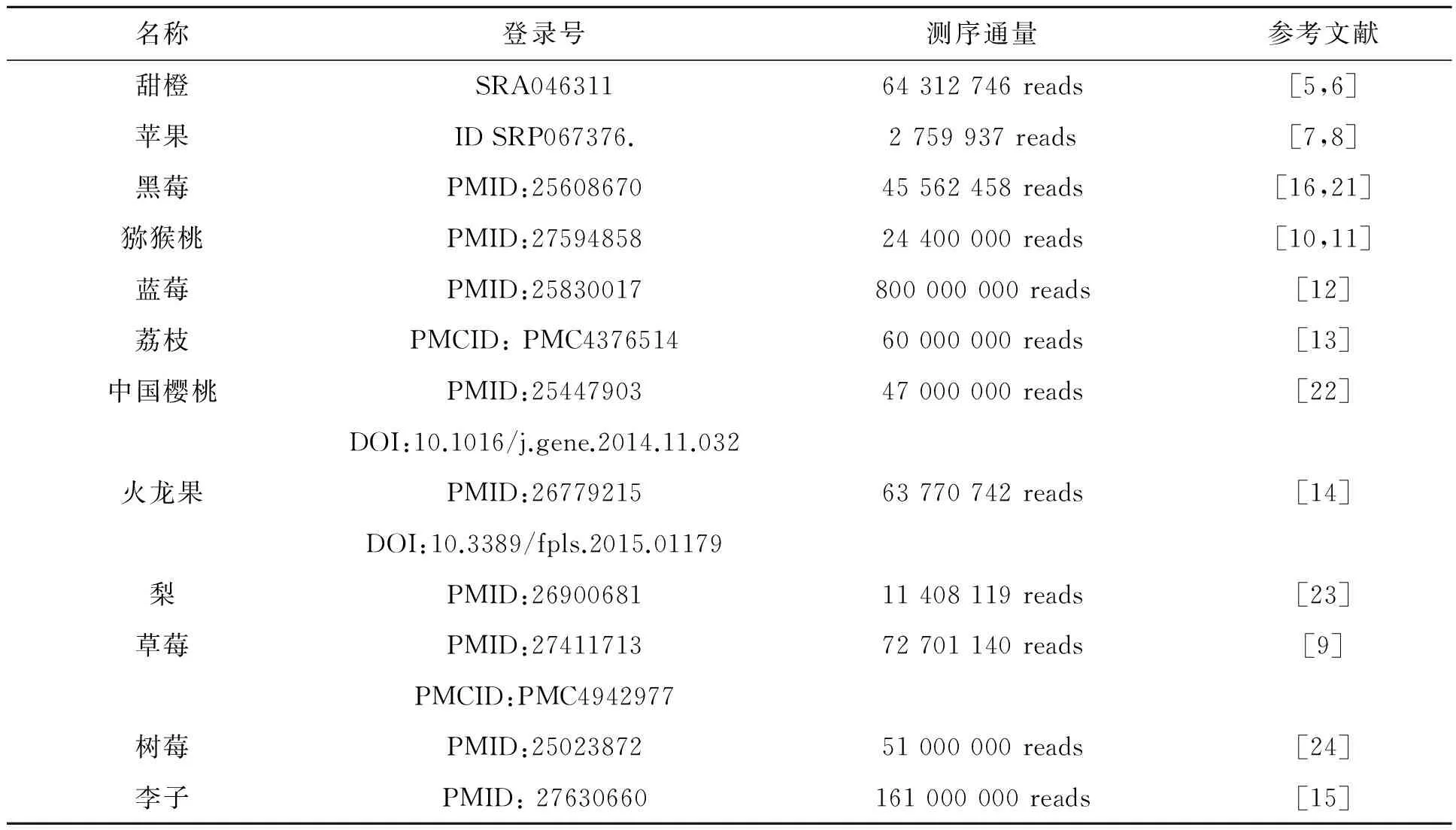
表1 应用RNA-Seq技术的部分果树Table 1 The trees used RNA-Seq.
RNA 高通量测序技术(RNA-Seq) 和单分子测序技术(SNS)[2]能提供精确的数字化信号,具有测序速度快、测序精度准确、测序成本低的特点,相比传统测序手段得到提升,将RNA-Seq结果与已知基因组DNA 序列信息进行对比,判断基因的表达情况、基因结构的优化以及新基因的发现等结果[20],是目前深入研究复杂转录组的强大工具。现已应用RNA-Seq 进行研究的部分果树信息见表1,得益于高通量测序技术的发展,使果树在分子生物学水平上的研究更为系统化,逐渐由分解转向整合[25],使木本植物型果树的分子生物学研究迎来新的机遇。
2 RNA-Seq技术在果树上的应用前景
2.1 基于RNA-Seq技术挖掘新基因
在Genbank上注册的基因序列都是经过与同源物种进行比对,或者通过EST测序预测而来的。如树莓(Rubuscorchorifolius)属于蔷薇科落叶小灌木,存在发育周期长、遗传背景复杂、基因组庞大等特点,单纯的基因注释结果往往难以完全涵盖某一物种的整个转录组信息。通过RNA-Seq技术,可以对现有的基因进行整合并优化现有的基因注释结果;通过测序数据与已知的参考基因组比对,能够发现一些新的转录本和新基因。
目前RNA-Seq技术在植物新基因的挖掘方面己有很多报道。应用Illuminate/Solexa测序平台对越橘果实的果肉组织进行转录组测序,共获得64 312 746个短序列[5],利用454测序平台对模式植物拟南芥的转录组进行重测序。Hyun等[24]第一次尝试使用Illumina测序技术在没有参考基因组的情况下,对红树莓进行转录组测序,共产生了5 100 000个短序列,经过重新组装,获得了42 604个平均长度为812 bp的独立基因,根据NCBI公共数据库比对,发现68.86%的独立基因在同源物种的植物中被发现。张晓慧等[10]利用RNA-Seq技术,探讨黄肉猕猴桃发育过程中果实类胡萝卜素合成途径中相关差异基因的功能发现:基因Unigene11266、Unigene23885 可能是类胡萝卜素合成的关键基因,且较其他基因而言,基因CL10467、Unigene12775、CL10798、CL1511、Unigene2673 与其关系更密切。
2.2 基于RNA-Seq技术确定次生代谢产物合成途径
针对一些具有经济价值或者药用价值的植物来说,一些重要次生代谢产物和有效成分的生物合成途径是研究的重点。由于果树体内次生代谢途径冗长而又复杂,因此利用RNA-Seq技术发现和挖掘这些重要途径中的关键酶基因为研究有效成分的生物合成途径和调控机制奠定了基础,同时为利用生物手段提高植物中的有效成分含量或直接生产有效成分提供了可能。基于高通量测序系统的RNA-Seq分析在植物次生代谢研究中具有重要意义。目前,已有多种植物通过转录组测序对其次生代谢途径进行了研究。Hyun等[24]对树莓进行转录组测序分析用于后续基因组功能和合成途径研究,和己知的蛋白质数据库NR、Swiss-Prot、KEGG和COG进行比对(E-value<1.0e″5),对其功能进行注释,在差异表达库中进行筛选,实验表明CHS、CHI、F3H、DFR、F3’H等7个关键酶基因与次生代谢产物花色苷合成相关。Gupta等[12]对蓝莓发育成熟前后5个阶段的转录组分析结果指出,蓝莓生长和成熟过程中涉及动态基因表达的变化,包括上调和下调代谢途径酶和转录调控因子。RNA序列比对分析确定了发育调控基因的选择性剪接过程及各阶段启动子的调控情况。最终得到了基因组序列、基因模型,并对其进行了GO功能注释。Petersen等[7]、Kui等[8]对苹果柱状苗的纯合子和杂合子分别进行RNA-Seq分析和RT-PCR分析验证,结果表明其初级根紧凑增长的分子基础已被确定,是由一个嵌套的gypsy-44反转录转座子插入;然而插入转座的表型以及时间之间的联系尚不清楚。Gerin等[26]利用RNA-Seq技术分析并揭示了黑曲霉菌OTA相关基因在转录过程的变化方式与途径,PKS催化异香豆素酸和L-苯丙氨酸之间的肽键的形成。
3 展望
尽管各测序平台在高通量水平、测序准确度、存储格式、技术方法上各有差异,但共同特征是大大降低了测序成本并极大地提高了测序速度。然而第二代测序技术需要将DNA从细胞中提取,然后将其片段化,再将接头连接到片段上,经PCR扩增后制成测序文库,但是在测序前PCR对待测片段进行扩增增加了测序的错误率。因此比较适合对已知序列的基因组进行重新测序,而在对全新的基因组进行测序时还需要结合第一代测序技术综合性地进行分析。对于树莓自身存在的发育周期长,遗传背景复杂、次生代谢产物合成途径受多种酶和底物限制,相对草本植物与动物的生物合成途径研究相对滞后,同时全基因测序进展缓慢等因素的影响。截至目前,该技术在果树上的应用还不健全,因此应用高通量测序技术手段完成树莓体内次生代谢产物的生命活动、代谢遗传规律进行系统化分析,从而揭示其生命活动的分子机理具有良好的前景。
[1] 白 波, 王春梅. 利用RNA-Sequencing 鉴定大鼠脑缺血再灌注损伤模型中差异表达的基因[J]. 济宁医学院学报, 2016, 39(1):18-22.
[2] 冯 超,朱长青,徐昌杰,等. RNA-Seq 在果树学研究中的应用[J]. 果树学报, 2014, 31(1):115-124.
[3] Parkinson J. Complementary DNA sequencing: expressed sequence tags and human genome projec[J]. Science, 1991, 252(3):1651-1656.
[4] 付 畅, 黄 宇. 转录组学平台技术及其在植物抗逆分子生物学中的应用[J]. 生物技术通报, 2011, 6(1):40-46.
[5] Wang J, Sun L, Xie L,etal.. Regulation of cuticle formation during fruit development andripening in ‘Newhall’ navel orange (Citrus sinensis Osbeck) revealed bytranscriptomic and metabolomic profiling[J]. Plant Sci., 2016, 243(3):131-144.
[6] Jonathan S, Zhou C. Transcriptome analysis of sweet orange trees infected with ‘CandidatusLiberibacterasiaticus’ and two strains of Citrus Tristeza virus[J]. BMC Genom., 2016, 17(1):349-361.
[7] Petersen R,Djozgic H. Johannes Columnar apple primary roots share some features of the columnar-specific gene expression profile of aerial plant parts as evidenced by RNA-Seq analysis[J]. BMC Plant Biol., 2015, 15(3):34-40.
[8] Kui L, Zhang J, Xie Y. Improved hybrid de novo genome assembly of domesticated apple (Malus×domestica)[J]. GigaScience, 2016,5(1):35-41.
[9] Zhang Y, Li W, Dou Y,etal.. Transcript quantification by RNA-Seq reveals differentially expressed henes in the red and yellow fruits ofFragariavesca[J]. PLoS ONE, 2015, 10(12):1-5.
[10] 张晓慧, 黄春辉, 钟 敏, 等. 基于RNA-seq 的黄肉猕猴桃类胡萝卜素合成相关基因的表达分析[J]. 果树学报,2015, 32(3):404-412.
[11] Wei T, Zheng Y, Dong J. Comprehensive transcriptome profiling reveals long noncoding RNA expression and alternative splicing regulation during fruit development and riening in kiwi fruit (Actinidiachinensis)[J]. Front Plant Sci., 2016, 7(3):335-342.
[12] Gupta V, Estrada A D, Blakley I. RNA-Seq analysis and annotation of a draft blueberry genome assembly identifies candidate genes involved in fruit ripening, biosynthesis of bioactive compounds, and stage-specific alternative splicing[J]. GigaScience, 2015, 4(1):5-11.
[13] Shu B, Li W, Liu L. Transcriptomes of arbuscular mycorrhizal fungi and litchi host inter action after tree girdling[J]. Front. Microbiol.,2016,5(3):1-14.
[14] Chengjie C, Zhe C, Pengkun C. Transcriptomic analysis reveals key genes related to betalain biosynthesis in pulp coloration ofHylocereuspolyrhizus[J]. Front. Plant Sci., 2016, 6(3):1-13.
[15] Fang Z, Zhou R, Fu Y. Identification of candidate anthocyan in-related genes by transcriptomic analysis of furongli plum (PrunussalicinaLindl.) during fruit ripening using RNA-Seq[J]. Front. Plant Sci., 2016, 7(3):13-38.
[16] Zhang Y, Martin C, Ramos B. RNA-Seq analysis and transcriptome assembly for blackberry (Rubussp. var.Lochness) fruit[J]. BMC Genom., 2015, 16(1):5-18.
[17] Hrdlickova R, Toloue M, Tian B. RNA-Seq methods for transcriptome analysis:RNA-Seq[J]. Radmila Hrdlicková, 2016, 10(3):13-24.
[18] 杨 帆, 黄立华, 张爱兵. 高通量转录组测序技术及其在鳞翅目昆虫上的应用[J]. 昆虫学报,2014, 57(1):991-1000.
[19] Zhang L,Zhou W. Characterization of the yeast transcriptome[J]. Cell, 1997, 88(2):243-251.
[20] Margulies A, Egholm W. Genome sequencing in microfabricated high-density picolitre reactors[J]. Nature, 2005, 437(7057):376-380.
[22] Zhu Y,Li Y,Xin D. RNA-Seq-based transcriptome analysis of dormant flower buds of Chinese cherry (Prunuspseudocerasus)[J]. Gene, 2015, 555(2):362-376.
[23] Li K, Xu X, Huang X. Identification of differentially expressed genes related to dehydration resistance in a highly drought-tolerant pear,Pyrusbetulaefolia, as through RNA-Seq[J]. PLoS ONE, 2016, 6(3):11-29.
[24] Hyun T, Lee S, Kumar D,etal.. RNA-Seq analysis of Rubus idaeus cv. Nova: transcriptome sequencing and de novo assembly for subsequent functional genomics approaches[J]. Plant Cell Rep., 2014, 33(10):1617-1628.
[25] 杨胜利. 系统生物学研究进展[J]. 中国科学院院刊, 2004, 19(1):31-34.
[26] Gerin D, Pollastro S, Faretra F. RNA-Seq reveals OTA-related gene transcriptional changes inAspergilluscarbonarius[J]. PLoS ONE, 2016, 11(1):1-21.
Progress on Application of RNA-Seq High Throughput Sequencing Technology in Functional Genomics of Fruit Trees
LIU Huan1, HUANG Lina2, Zhang Fenqiang1, JIANG Mengyan2, ZHOU Yijie1, CHEN Siyuan2, WANG Younian1, YANG Mingfeng2*, MA Lanqing2,3*
1.KeyLaboratoryofUrbanAgriculture(NorthChina),MinistryofAgriculture,PlantScienceandTechnologyCollege,BeijingUniversityofAgriculture,Beijing102206,China;2.KeyLaboratoryofUrbanAgriculture(NorthChina),MinistryofAgriculture,CollegeofBiologicalScienceandEngineering,BeijingUniversityofAgriculture,Beijing102206,China;3.BeijingCollaborativeInnovationCenterforEco-environmentalImprovementwithForestryandFruitTrees,BeijingUniversityofAgriculture,Beijing102206,China
RNA-Seq, also called "Transcriptome Sequencing Technology", can be used to study the species gene function and genetic structure at an overall level to reveal specific biological processes. The application of this technique can effectively resolve the issue of huge genome of fruit trees and other problems, and efficaciously reveal and elucidate biosynthetic pathways and metabolic mechanisms of secondary metabolism in the plant. This article mainly discussed the application method and processes of the RNA-Seq technology; We analized in-depth the synthesis mechanisms of rich small molecular compounds with various secondary bioactive metabolites in synthetic fruit trees and carried out researches on the key enzyme genes synthesizing this pathway. The high-throughput sequencing technology based on RNA-Seq makes it possible to discover and reveal the key genes and regulation and control mechanisms for the synthesis of secondary metabolites with important biological functions and economic values.
high throughput sequencing; RNA-Seq; fruit tree; secondary metabolite
2016-11-17; 接受日期:2017-01-12
国家自然科学基金项目(21606020);北京市自然科学基金项目(2164059);北京市科技创新服务能力建设-协同创新中心项目(PXM2017_014207_000043)资助。
刘欢,硕士研究生,研究方向为果树品质优质生态安全。E-mail:huanliu0824@163.com。*通信作者:杨明峰,副教授,研究方向为植物分子生物学和植物代谢调控研究。E-mail:mingfengyang@bua.edu.cn。马兰青,教授,研究方向为植物次生代谢分子调控。E-mail:lqma713@163.com
10.3969/j.issn.2095-2341.2017.02.11
