分子模拟方法研究四氢化吡啶并[1,2-a]吲哚酮衍生物对GSK3β和CDK5的选择性
2016-11-28董珂珂杨雪雨赵腾腾朱小蕾
董珂珂 杨雪雨 赵腾腾 朱小蕾
(南京工业大学化工学院,材料化学工程国家重点实验室,南京210009)
分子模拟方法研究四氢化吡啶并[1,2-a]吲哚酮衍生物对GSK3β和CDK5的选择性
董珂珂杨雪雨赵腾腾朱小蕾*
(南京工业大学化工学院,材料化学工程国家重点实验室,南京210009)
本文通过分子对接,分子动力学模拟(MD)和MM/PBSA能量计算的方法,从分子水平研究了3个四氢化吡啶并[1,2-a]吲哚酮衍生物与CDK5和GSK3β的相互作用,并揭示了这些抑制剂对GSK3β的选择性抑制机理。分子对接结果表明,抑制剂对2种激酶具有相似的结合模式,结合口袋处的残基也都根据晶体结构的序列比对相互对应。研究体系的RMSD随时间的稳定变化,表明模拟体系已达到稳定状态,因而后续的分析是可靠的。CDK5/抑制剂体系,RMSD在0.15 nm上下波动,CDK5/M1和CDK5/M2骨架轻微波动,稍高于CDK5/M3;而GSK3β体系的RMSD值略高于CDK5体系,在0.17 nm上下波动,GSK3β/M1和GSK3ββ/M2的骨架波动平衡值则稍低于GSK3β/M3。活性较大的抑制剂增强了蛋白骨架整体的“柔性”,即对激酶构象产生一定影响。能量分析表明,静电能和范德华作用能够区分不同抑制剂对同种激酶的生物活性差异。极性溶剂化自由能对区分抑制剂选择性也很重要,残基分解表明GSK3β的Glu97、Thr138是造成抑制剂选择性的主要原因。抑制剂与CDK5和GSK3β结合的过程中,蛋白质残基的动态相关性存在差异,铰链区域的Thr138与Val135~Gln206区域残基正相关,证实Thr138残基是区分抑制剂选择性的关键。
四氢化吡啶并[1,2-a]吲哚酮衍生物;分子动力学模拟;选择性;稳定性
许多人类疾病通常都是由蛋白异常磷酸化所引起的。人类基因组中包含518种蛋白激酶,其中细胞周期素依赖性蛋白激酶(CDKs)和糖原合成激酶-3(GSK3)都属于丝氨酸/苏氨酸蛋白激酶家族[1],由于两种激酶在大多数病理学中频繁出现的反常现象而引起关注。CDKs包含一个特定的丝氨酸/苏氨酸催化位点,与调控分子的周期蛋白亚基一起控制激酶活性和亚基的特异性,能够调控细胞周期循环的G1期、M期、S期、G2期的相互转换以及影响其他酶的转录、复制,促进细胞分裂[2-4],因此在细胞循环中起着重要作用。GSK3激酶由两种单独的基因编译分别得到-α和-β两种形式的激酶,通过磷酸化多种蛋白酶,参与多种生物学过程如干细胞自我更新、细胞分裂、细胞分化、细胞凋亡、生理节奏、转录和胰岛素作用,其反常调节导致了许多代谢紊乱,神经系统疾病(如双向性精神障碍、精神分裂症和阿尔兹海默病(AD)等)以及癌症的发生[5-8]。GSK3的两种亚型激酶结构相似,但作用不同,GSK3活性的抑制通过保守性丝氨酸残基GSK3α的Ser21和GSK3β的Ser9的磷酸化控制[9],在ATP竞争性结合位点处也有氨基酸的差异(GSK3α的Glu196和GSK3β的Asp133)。
然而,神经系统疾病中,阿尔兹海默病是痴呆的最为普遍的形式,世界卫生组织报道2015年,全世界近480万人患上AD病,其主要病理为由tau蛋白过度磷酸化导致的神经元纤维缠结[10],β淀粉样多肽(Aβ)沉积导致的老年斑等有毒特性以及由神经元突触和整个神经元缺失引起的脑萎缩[7]。而GSK3β的活化结构在AD患者中被发现,与聚集成双螺旋丝的tau蛋白中tau的过度磷酸化直接相关[11]。当GSK3β被激活时,阿尔兹海默病患者的记忆能力衰退,影响突触可塑性,损伤突触的形态和功能[12],同时还参与胞内和胞外通道,包括wnt信号和胰岛素信号通道,以及G-蛋白偶联受体和其他蛋白酶,主要通过磷酸化影响。另外,CDK5,作为CDKs家族的一员,不同于其他细胞周期素依赖性激酶涉及到细胞的增殖,细胞分裂等,其与激活因子p35和p39结合在神经元中达到高的活性,与通过钙蛋白酶剪切得到的p25结合后导致CDK5的过度活化,导致AD病的发生;而相反CDK5的缺失则会抑制过度兴奋,减少焦虑和记忆损伤[13];CDK5与GSK3β类似在神经系统,突触信号传导以及学习和记忆过程中发挥重要作用[14]。几个引起AD的因素如发炎和氧化应激也能够导致CDK5活性增加。总的说来,这些研究都强有力地表明CDK5与GSK3β在许多神经类疾病中都起着重要作用,目前都已成为治疗多种病症的重要靶标和新药开发的热点。
由于CDKs与GSK3β的结构相似性,不难发现大多数GSK3β的抑制剂同样抑制CDKs,如Indirubin(靛玉红类)[15]、Aloisines[16]、Hymenialdisine (生物碱类)[17]等。配体诱导的靶标蛋白来调控其活性在癌症中已经成为有效的治疗策略,然而受限于许多因素包括较差的靶向特异性和低的抑制潜能,选择具有选择性抑制剂能够克服药物抗性,减少毒副作用等[18]。目前研究的GSK3β而非CDK5的选择性抑制剂有:Pyrazines(吡嗪类)[19]、Paullones(苯醌类)[20]和Valmerins(吡啶并吲哚酮类)[21-22]等。由于对GSK3β的选择性机理并不明确也使得选择性抑制剂的开发变得缓慢,并且实验无法从分子水平详细解释,这就需要通过计算来解决。Dessalew等[23]用3D-QSAR和分子对接的方法研究了Bisarylmaleimide(二苯马来酰亚胺)对于GSK3β的选择性抑制,而对CDK2和CDK4活性较弱,通过打分评价了选择性的定量差异以及与实验的一致性。Chen等[20]用动力学模拟以及能量计算的方法研究了苯醌类抑制剂对于GSK3的选择,计算表明了该抑制剂与GSK3的Val135残基比与CDK5的Cys83残基有更强的相互作用,同时与CDK5的Asp86残基的相互作用较弱,导致了抑制剂的特异性,结构间的差异分析较少。然而吡啶并吲哚酮类抑制剂对GSK3β有较大的选择性,但机理并不清楚。
本文用分子对接、分子动力学模拟、MM/PBSA (GBSA)能量计算,以及残基间相互作用分析的方法首次深入研究由Rajaa Boulahjar合成的四氢化吡啶并[1,2-a]吲哚酮衍生物[21]为什么对GSK3β抑制,而对结构和功能相似的CDK5却有较低的抑制活性。这对于潜在的结构修饰以及开发更具潜力和选择性的GSK3β的吡啶并吲哚酮抑制剂提供深入的指导。
1 实验部分
1.1配体和受体结构准备
利用3D画图软件构建3个四氢化吡啶并[1,2-a]吲哚酮衍生物(图1),并在B3LYP/6-31G(d)水平上用Gaussian 09软件[24]进行优化得到最优结构。通过Amber软件[25]的Antechamber模块拟合限制性静电势得到原子电荷以及其他的力场参数等,这种方法在许多文献[26-28]中被证明是有效的。CDK5(Met1~Pro292)和GSK-3β(Ser35~Ile384)的晶体结构从蛋白质数据库(the protein data bank)得到,PDB ID分别为1UNL[29]和2O5K[30],分辨率为0.20 nm,删除其中的水分子和抑制剂分子。CDK5的晶体结构为CDK5/p25二聚体,因此仅选取A链的CDK5和D链的p25作为对接的初始结构,以下简写为CDK5。
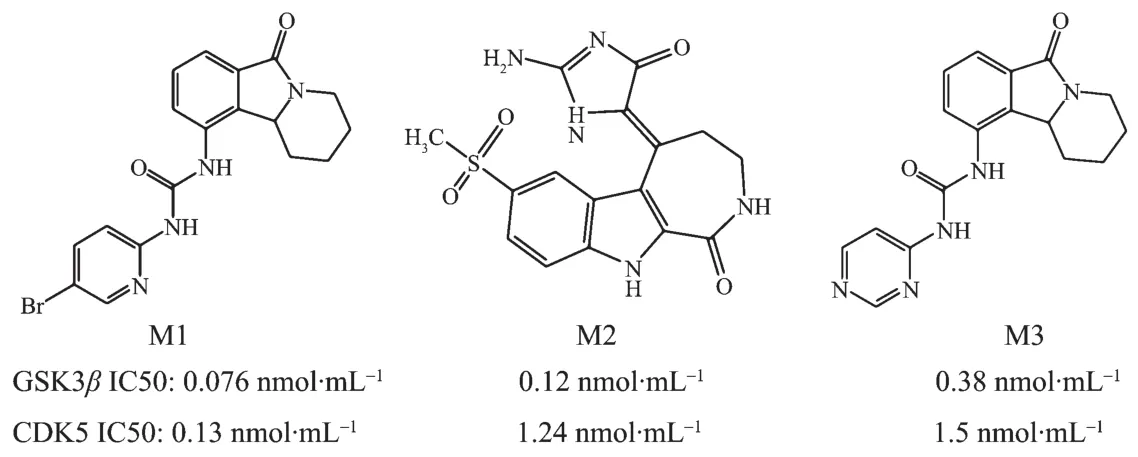
图1 3个GSK3β选择性抑制剂的结构和IC50[21]值Fig.1 Structures and in vitro activity of three specific inhibitors of GSK3β
1.2分子对接
小分子配体与受体的相互作用首先通过分子对接研究。分子对接采用Autodock4.0程序的半柔性对接方法,将极性氢原子添加到蛋白质的各个残基上,配体的键可自由旋转,受体则为刚性。将格点盒子的中心设在两种激酶的ATP竞争性结合位点处,以使配体对接到此处,格点盒子大小设为90× 90×90,间隔为0.037 5 nm,用遗传算法共进行200次独立对接计算,其他参数默认,根据均方根偏差(RMSD)在0.20 nm范围的标准聚类分析,得到最优构象。且分别与含有R-roscovitine抑制剂的CDK5复合物(1UNL),和含有抑制剂GSK3β的复合物比对,RMSD值都在0.10 nm内,因此证明对接结构也是合理的,为后续的MD分析奠定基础。
1.3分子动力学模拟
在分子对接的基础上,3个小分子抑制剂与两种激酶形成的6个复合物体系采用Amber10[25]的Sander和Pmemd模块进行MD模拟。与许多研究者[20,23,31-33]一样,本文配体和受体分别采用GAFF力场[34]和AMBERFF03力场[35],每个体系都融入被截断的八面体的TIP3P水分子类型[36]的水盒子中,复合物与盒子边界的最短距离为1 nm。氢原子通过LEaP模块添加,残基的质子化状态在中性的pH值下采用默认,并添加抗衡离子(Na+或Cl-)使体系电荷呈中性。每个体系的能量最小化采用2 000步的最小化法和2 000步的共轭梯度法,非键截断半径设为1 nm。MD模拟过程主要包括200 ps的41 840 kJ·mol-1·nm-2)限制性加热(0~300 K),200 ps的密度平衡,200 ps的平衡以及在NPT系综(P=101 kPa,T=300 K)下,时间步长为0.002 ps的20 ns未加限制的MD模拟。通过SHAKE算法[37]对氢原子在内的所有键固定,用Particle-mesh Ewald(PME)方法[38]处理长程静电相互作用。MD轨迹坐标每1 ps保存一次用于每个体系的结构和能量分析。
1.4结合自由能计算
分子动力学模拟之后,通过每个体系的骨架原子的RMSD来评价其稳定性。然后通过MM/PBSA (GBSA)方法[39],取稳定后的5 ns的轨迹,提取200个构象对抑制剂与两种激酶的结合自由能计算来研究结合亲和力的差异以及相同抑制剂与不同激酶的能量差异研究抑制剂选择性。每个快照(snapshot),配体与受体结合自由能差(ΔGbind)可表述为:

其中ΔGcomplex、ΔGprotein和ΔGligand)分别代表复合物、蛋白质以及配体的自由能。结合自由能又可分解为三部分:气相结合能(ΔEgas)、溶剂化自由能(ΔGsol)和熵贡献(TΔS)三部分(方程2)。ΔEgas又可进一步分解成静电能(ΔEele)和范德华作用能(ΔEvdw)(方程3)。溶剂化自由能(ΔGsol)可以分为两部分:极性溶剂化自由能(ΔGpolar)和非极性溶剂化自由能(ΔGnonpolar)(方程4)。极性溶剂化自由能通过amber10中的pbsa模块解线性的PB(Poisson-Boltzmann)方程得到,溶剂探针半径设为0.14 nm。溶质和溶剂的介电常数分别设为1和80。通过用Molsurf方法,可以得到溶剂可及表面积(SASA),将溶剂参数γ和β分别设为0.226 77 kJ·mol-1·nm-1)和3.85 kJ·mol-1来计算非极性溶剂化自由能(ΔGnonpolar)(方程5)。由于抑制剂的选择性主要在于CDK5和GSK3β之间的相似度,以及结合位点附近的残基的差异。通过Clustal omega[24]软件我们得到两个序列的最佳比对,两种激酶的序列一致性为29.0%,相似度为45.4%。另一方面,抑制剂对激酶的选择性差异也在于两种激酶相似位置的残基对极性溶剂化自由能的贡献差值,可以表述为(方程6)。一般说来,对于较大的体系计算熵非常耗时,并且对于相似的受体来说小分子的结合导致的熵变很接近[40-41],为减少计算成本,许多文献在计算过程都忽略了熵变[42-44],本文我们也忽略熵变。
1.5动态相关性分析
为了评价不同蛋白区域之间的动态相关性,对3个抑制剂分别与CDK5和GSK3β结合的6个体系进行了动态相关性Dynamic Cross-Correlation Map(DCCM)分析[45-46]。两个原子i和j之间的相关系数Cij定义为:

其中ΔRi表示第i个原子相对于它的平均位置的瞬时波动。正相关表示残基以相同的方向运动,Cij=1;负相关表示残基以相反方向运动,Cij=-1。整个20 ns模拟过程中,最低能量结构用于DCCM分析。PyMol v1.8软件[47]用于蛋白质结构作图分析。
2 结果与讨论
2.1对接构象结合模式
3个四氢化吡啶并[1,2-a]吲哚酮衍生物抑制剂对接到CDK5和GSK3β的ATP竞争性结合口袋中(图2),发现两者具有相似的折叠结构,且结合位点也非常相似,都有一个很大的空腔,由周围残基(CDK5:I10、C83、L133和N144;GSK3β:I62、V135、L188和D200)包围,主链原子叠合得都很好,与晶体结构的序列比对一致[20]。由图2可知,在3个抑制剂与CDK5的对接结构中,3个抑制剂都处于由残基Ile10、Glu12、Gly13、Val18、Ala31、Glu81、Phe82、Cys83、Asp84、Asp86、Gln130、Leu133和Asn144组成的疏水口袋中。与之相比,与GSK3β对接的复合物结构中,3个抑制剂位于由Ile62、Val70、Ala83、Asp133、Tyr134、Val135、Pro136、Gln185、Asn186、Leu188和Asp200组成的疏水性口袋中,这与其他文献的对接研究结果一致[48]。值得注意的是,CDK5与M1和M2的相互作用残基都有Lys33、Gln85、Lys89,而CDK5/M3复合物中则没有,另外,小分子M1周围的CDK5残基数目相比M2和M3也是最多的。说明这几个残基对于抑制剂与激酶的结合比较关键,相应地,GSK3β的Lys85、Glu137、Arg141残基也有此功能。
2.2体系的稳定性和柔性分析
由对接构象可知,抑制剂与激酶之间形成的氢键作用使得复合物体系更加稳定。结合抑制剂不同结构构象也有差异,且抑制剂与激酶结合过程也是动态的,这也造成了动力学特性包括稳定性和柔性发生变化,结合自由能也由于抑制剂活性与选择性的不同而不同。因此,本文对3个抑制剂与CDK5和GSK3β结合的6个体系在300 K下进行了20 ns的分子动力学模拟。在15 ns后,6个体系均达到平衡(图3)。因而后续的能量和结构分析是可靠的。对CDK5/抑制剂体系,RMSD在0.15 nm上下波动,CDK5/M1和CDK5/M2骨架轻微波动,稍高于CDK5/M3,表明活性较大的抑制剂增强了蛋白骨架的“柔性”;而对GSK3β体系,RMSD在0.17 nm上下波动,GSK3β/M1和GSK3β/M2的骨架波动平衡值则稍低于GSK3β/M3。这表明3个抑制剂对2个激酶体系的活性差异有着不同机理。

图2 CDK5/抑制剂(蓝色)和GSK3β/抑制剂(红色)对接后结构中ATP结合口袋的叠加Fig.2 Superimposition of the ATP-binding pocket of the CDK5/inhibitor(blue)and GSK3β/inhibitor(red)structures after dock
蛋白质柔性也由平均结构的均方根波动(RMSF)来评判,通过RMSF值也可以判断MD模拟过程是否收敛。本文我们计算了6个体系的蛋白质骨架原子上的RMSF值,结果如图4所示。总体上,所有体系的蛋白质结构与实验晶体结构的RMSF值(由B-factor换算得到)分布基本一致。两种蛋白质柔性略有差异,而3个抑制剂与两种激酶的结合位点处的RMSF值相比几乎没变化,都略小于实验中CDK5的RMSF值,表明活性区域有较高的刚性,活性越大,刚性也越强。GSK3β结构符合观察到的“活化片段”蛋白激酶,包含一个N端的β-sheet区域和一个C端的α-helical区域。7个反平行的β折叠形成一个封闭的正交桶状结构,即N端(Ser35~Tyr134),其中在3个抑制剂与GSK3β结合的复合物中较为显著的β5和β6连接的loop区域(Ser119~Glu125),因其活性不同表现了不同的柔性,GSK3β/M3的RMSF值最高,柔性最大;参与催化活化区域(Gly210~Arg220)无论磷酸化与否,结构都处于活化状态[49],从图4中也可以看出与3个抑制剂结合都表现了很强的刚性;结构可塑性延伸的loop区域(Asn285~His299)的柔性变化也与抑制剂活性相关,正如Dajani等[50]研究的axin和FRAT抑制剂与GSK3β结合造成的柔性不同,我们发现活性越大,刚性越强。侧链残基Ile62(G-loop区域)和Hinge区域的Val135残基的柔性变化都不大。这些区域对于抑制剂的选择性也有一定的影响。抑制剂翻转或者其他构象改变引起的范德华或静电作用将对结合有一定影响。

图3 GSK3β和CDK5两激酶与3个抑制剂形成的复合物的RMSD值随时间的变化Fig.3 RMSD of backbone atoms of two kinases(GSK3β and CDK5)with three inhibitors as a function of time

图4 CDK5(左)和GSK3β(右)的每个残基主链原子对应的RMSF变化Fig.4 Root-mean-squared fluctuation(RMSF)of the backbone atoms of each amino acid residue of CDK5(left)and GSK3β(right)
回旋半径是蛋白质致密性(compactness)的一个指标[51-52],为了更直观地从整体上了解抑制剂对蛋白质分子尺寸的影响,本文通过回旋半径(Radius of Gyration,Rg)的计算研究了CDK5和GSK3β两种激酶残基的致密程度。CDK5/抑制剂复合物的回旋半径平均值为2.364 nm±0.005 nm,这与前人计算得到的CDK5/p25/roscovitine复合物体系的Rg(2.384 nm±0.008 nm),CDK5/p25的Rg(2.360 nm±0.008 nm)较一致,同时与实验结果(Rg(CDK5/p25/roscovitine):2.347 nm)[53]也没有很大差异。GSK3β/抑制剂体系的Rg平均值为2.145 nm±0.008 nm,3个体系Rg值较为类似,但稍小于CDK5体系,这也是由于CDK5/ p25蛋白质残基数目较多导致的。结果表明两种激酶体系并未因为抑制剂的结合而导致蛋白质残基的致密性发生变化,模拟过程中蛋白也是稳定的。
2.3抑制剂对CDK5和GSK3β的生物活性
抑制剂与激酶的结合自由能计算是衡量抑制剂活性的重要方法,且计算得到的结合自由能与实验活性值比较更进一步判断计算的适用性和准确性。MM/PBSA方法和MM/GBSA方法是计算结合自由能的常用方法,许多研究工作者用MM/PBSA的方法计算结合自由能[39,54-55],且研究证明MM/PBSA方法比MM/GBSA方法更加准确[54]。本文我们用MM/PBSA的方法计算了6个复合物体系的结合自由能以及不同能量项,结果列在表1中。表1中的能量值,正数表示这种作用不利于抑制剂与激酶的结合,负数表示这种作用使体系能量降低,有利于结合,负数绝对值越大,表示这种作用对结合的贡献越大。
M1~M3对CDK5的结合自由能分别为:-107.57、-89.75、-68.95 kJ·mol-1,与实验中抑制剂生物活性(IC50值分别为0.13、1.24、1.5 nmol·mL-1)一致;对GSK3β的结合自由能分别为:-109.04、-92.72、-91.88 kJ·mol-1,同样与实验中的抑制剂抑制活性(IC50值分别为0.076、0.12、0.38 nmol·mL-1) (图1)[21]相一致。同时3个抑制剂也表现出了对GSK3β的选择性。为了深入了解哪部分相互作用引起了抑制剂与2种激酶的结合,我们分析了各个能量项对结合亲和力的贡献大小。如表1所示,静电能(ΔEele)、范德华作用项(ΔEvdW)和非极性溶剂化自由能(ΔGnonpolar)对结合是有利的。6个体系中,范德华作用都起主导作用。对于GSK3β体系,范德华作用能区分3个抑制剂的生物活性,而对CDK5体系,CDK5/M2和CDK5/M3的极性溶剂化自由能可区分其生物活性。尽管气相的静电能(ΔEele)对结合是有利的,但也不能完全抵消极性溶剂化自由能(ΔGpolar)对结合的不利影响。所有体系的非极性溶剂化自由能差别都不大。

表1 用MM-PBSA方法计算得到的蛋白质-配体复合物的结合自由能项aTable1 Binding free energy components for the proteininhibitor complexes by using the MM-PBSA methoda
为了深入地研究蛋白质的每个残基在抑制剂的结合中发挥的作用,我们对每个残基进行了能量分解。6个体系分解到每个残基的结合自由能如图6和图9所示。图6中我们列出了能量小于-4.184 kJ·mol-1的残基,M1与GSK3β的结合亲和力主要作用残基为I62、Val70、Lys85、Thr138、Leu188和Cys199,M3的关键残基与M1的类似,而M2的关键残基则略有不同,Lys85残基贡献变小,这可能是因为M1与M3结构(用嘧啶基取代M1中的3-溴吡啶基)较为相似,而与M2骨架有较大差别有关。M2的可旋转键较少,有极性基团的甲基磺酰基,尽管磺酰基和羰基大小相似,并有相似的电荷分布,但其提供2个氢键受体,在复合物中,伸向带正电荷极性Lys85残基(图7),两者间作用力则较弱。另外,Lys85残基对3个抑制剂结合的贡献大小在能量上也体现在静电作用,其静电能分别为:-38.79、-14.14、-33.39 kJ·mol-1,这个差异主要是抑制剂骨架结构不同造成的。
由上述总的自由能和自由能分解项可知,范德华作用主要区分抑制剂对GSK3β的生物活性,因此我们计算了每个残基的范德华作用能贡献,如图8所示,GSK3β/M1~M3复合物针对范德华作用贡献的关键残基由活性的减小而减小,计算所得与实验测得的生物活性相符。GSK3β/M1的关键残基为:Ile62、Val70、Tyr134、Thr138、Arg141、Leu188和Cys199,与两外两种复合物相比,作用主要集中在残基Ile62、Val70、Leu188和Cys199。与总的结合能残基分解相一致。
我们研究的3个抑制剂对CDK5和GSK3β两种激酶都有选择性,CDK5/M1复合物体系对能量的残基分解显示的关键残基要少于GSK3β/M1体系,主要表现在残基:Gly11、Val18、Phe80、Gln130、Asn131、Leu133和Asn144(图9),其中Val18和Leu133几乎与GSK3β/M1的Val70和Leu188残基发挥同样的作用。CDK5/M1与CDK5/M3的关键残基相似,说明有相似的相互作用,而CDK5/M2中Ile10残基还对结合有较大的贡献,其他2种复合物则没有。另外Lys33残基在CDK5/M1中起着不利作用,对应于GSK3β中的Lys85残基,是区分选择性的关键性残基。

图6 抑制剂(M1、M2和M3)与GSK3β的每个残基之间的相互作用能Fig.6 Interaction energies between inhibitors(M1,M2 and M3)and each individual residue of GSK3β
图7 3个复合物MD后最低能量结构构象图Fig.7 Conformations with lowest energy after MD simulations for three complexes

图8 抑制剂(M1、M2和M3)与GSK3β复合物抑制剂-残基的范德华作用Fig.8 Van der Waals interaction energy of inhibitor-residue pair in GSK3β/inhibitor complexes
由上述图1和表1的能量项分解可知,尽管M1和M3有着较小的差别,其静电能和范德华能都能够区分其生物活性。对于M1和M2来说,结构骨架差异较大,但也能通过这两项来区分。对于结构极为相似的抑制剂M1和M3,其范德华作用主要残基也是相似的(图10),其中主要区分其生物活性的残基是:Asn131和Asn144。这与后续的氢键分析一致,同时与M1和M3两个抑制剂的结构差异有关(M1中的3-溴吡啶基和M3中的嘧啶基的差异)。
前面,我们从能量的角度揭示了静电作用和范德华作用如何影响抑制剂对激酶的生物活性。下面我们将更详细地分析这些作用对抑制剂的生物活性的影响。如图11所示,我们用Ligplot+程序[56]讨论2种激酶和抑制剂的结合模式。CDK5/M1体系中,M1与相邻残基形成了2个稳定的氢键。一个形成于M1的连接四氢吡啶并吲哚酮大环和吡啶环的骨架氮原子(N14)与Gln130的氧原子之间,另一个形成于M1中的连接3-溴吡啶基的氮原子(N15)与Gln130的氧原子之间;整个模拟过程中氢键占有率分别为98.15%和96.62%,键长平均为0.298和0.289 nm,且是稳定存在的(图12)。此外,M1还与周围残基形成了疏水作用,3-溴吡啶基与Glu12、Gln130和Asn144残基之间的疏水作用;四氢吡啶并吲哚酮大环则与Ile10、Val18、Ala31、Asp86和Leu133残基形成疏水作用。而GSK3β的更多的残基与M1产生疏水作用,3-溴吡啶基与Ile62、Tyr134、Pro136和Arg141;四氢吡啶并吲哚酮大环与Val70、Ala83、Lys85、Leu132、Tyr134、Pro136、Gln185、Asn186、Leu188和Cys199残基形成的疏水作用,发现相同基团产生的疏水作用却是不同的,表明M1与GSK3β结合时发生了翻转,且对GSK3β表现出了选择性。
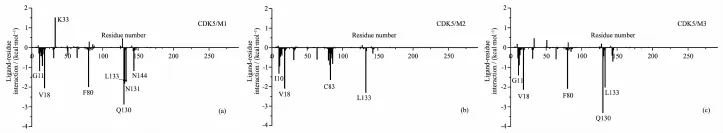
图9 抑制剂(M1、M2和M3)与CDK5的每个残基之间的相互作用能Fig.9 Interaction energies between inhibitors(M1,M2 and M3)and each individual residue of CDK5

图10 抑制剂(M1和M3)与CDK5复合物抑制剂-残基的范德华作用Fig.10 Van der Waals interaction energy of inhibitor-residue pair in CDK5/inhibitor complexes

图11 氢键和疏水作用二维图Fig.11 2D representation of hydrogen bond and hydrophobic interaction
M2与CDK5形成了3个氢键,其中2个是与咪唑基相连的羰基氧原子(O20)和氮原子(N21)分别与Cys83残基的N原子和Asp86残基上的氧原子(OD1)之间形成的氢键(表2)。另外1个则是磺酰基的氧原子(O24)与Glu12上的氧原子形成的氢键,LigPlot+的氢键二维图显示M2与GSK3β也形成3个氢键,与M2有疏水作用的残基相比CDK5来说也是增多的。M3与2种激酶的疏水作用残基数目一样,M3的嘧啶环与CDK5中的Gly13、Lys33、 Phe80、Asn131和Asn144有疏水作用,而在GSK3β中则是Ile62、Tyr134、Val135、Pro136和Arg141残基,与CDK5中残基Lys33序列比对的GSK3β中的残基Lys85与M3形成了氢键,尽管氢键占有率较低,但最终是稳定的。抑制剂与2种激酶的结合模式分析表明,GSK3β中的Lys85残基对于区分选择性比较关键。
2.4抑制剂对GSK3β的选择性
基于上述能量分解,我们发现极性溶剂化自由能能够区分抑制剂对于GSK3β的选择性。尽管极性溶剂化自由能对结合是不利的,但对比CDK5和GSK3β两种激酶与抑制剂的复合物发现,CDK5与抑制剂之间的极性溶剂化自由能对结合更加不利。这是由两种激酶结构差异导致的极性溶剂化自由能的差异,而这体现了四氢化吡啶并[1,2-a]吲哚酮衍生物对GSK3β的选择性。

图12 抑制剂与CDK5之间形成的H键的距离随MD模拟的时间变化Fig.12 Interatomic distances in the course of the MD simulations show the stability of formed hydrogen bonds between three inhibitors(M1,M2 and M3)and CDK5

表2 抑制剂与CDK5和GSK3β之间形成的氢键Table2 Hydrogen bonds formed between inhibitors and CDK5 or GSK3β
基于2个激酶的序列比对,我们通过比对残基的极性溶剂化自由能贡献的差值来研究抑制剂的选择性,如图13所示。纵坐标为了能量间隔显示一致,对于M2抑制剂,GSK3β的Thr138与CDK5的Gln85残基的差值被截断,实际值为-39.75 kJ· mol-1。M1~M3比较,从图中不难发现,GSK3β的Glu97、Thr138是造成抑制剂选择性的主要原因,而Lys85则对于抑制剂对GSK3β的选择性不利。这与前边活性和结构差异的讨论一致。那么通过减小抑制剂对GSK3β中的Glu97和Thr138的不利作用,即增强抑制剂极性,理论上可以提高抑制剂对GSK3β的特异性。
通过上述能量分析我们了解到抑制剂与两种激酶的相互作用机制以及抑制剂对GSK3β的选择性机制,而在抑制剂与同源性很强的CDK5和GSK3β结合的过程中,其对两种激酶本身结构,即激酶中各残基间的相互作用影响的差异也是造成抑制剂选择性的重要因素。为此我们针对M1采用了动态相关性分析(DCCM),如图14所示。M1与两种激酶结合时,CDK5的Ile10~Arg36残基与GSK3β的Ile62~Val70的相关性比较类似,差别较大的则是Val135~Val272区域蛋白质残基的相关性,这些残基所对应的CDK5的区域则是Cys83~Leu217残基。对于GSK3β的这部分区域几乎都是正相关的,蛋白质残基运动方向一致,包括铰链(Hinge)区域的Val135和Thr138与Val135~Gln206区域残基的运动一致。而对应于CDK5的这部分区域则不显著。这从蛋白质结构本身也说明了GSK3β的Thr138残基在筛选CDK5和GSK3β的选择性抑制剂中的重要性。

图13 GSK3β和CDK5之间的相对每个残基对的极性溶剂化自由能的差异Fig.13 Difference in the polar solvation energies ΔΔGpolbetween GSK3β and CDK5 as a function of residue index

图14 GSK3β/M1和CDK5/M1的动态相关性(DCCM)分析Fig.14 Dynamic cross-correlation map(DCCM)analyses for GSK3β/M1 and CDK5/M1
3 结论
本文通过分子对接,分子动力学模拟(MD)、MM/ PBSA能量计算以及蛋白质结构的分析相结合的方法,从分子水平研究了3个四氢化吡啶并[1,2-a]吲哚酮衍生物与CDK5和GSK3β的相互作用,并揭示了该抑制剂对GSK3β的选择性抑制机理。分子对接结果表明,抑制剂对2种激酶具有相似的结合模式,结合口袋处的残基也都根据序列比对相互对应。GSK3β体系的平均RMSD值(0.17 nm)略高于CDK5体系(0.15 nm),且活性较大的抑制剂RMSD值较低,因为活性较大的抑制剂增强了蛋白骨架整体的“柔性”,即对激酶构象产生影响一定影响。能量分析表明,静电能和范德华作用能够区分不同抑制剂对同种激酶的生物活性差异。极性溶剂化自由能对区分抑制剂选择性很重要,残基分解表明GSK3β的Glu97、Thr138是造成抑制剂选择性的主要原因。而CDK5和GSK3β在与抑制剂结合过程蛋白质残基的动态相关性也存在差异,铰链区域的Thr138与Val135~Gln206区域残基正相关,证实Thr138残基是区分抑制剂选择性的关键。本文采用分子动力学模拟的方法从结构和能量方面较好地解释了四氢化吡啶并[1,2-a]吲哚酮衍生物对GSK3β的选择性抑制机理,为改善GSK3β选择性抑制剂提供数据和理论指导。
[1]Frame S,Cohen P.Biochem.J.,2001,359:1-16
[2]Nurse P,Masui Y,Hartwell L.Nat.Med.,1998,4:1103-1106
[3]Sherr C J.Science,1996,274:1672-1677
[4]Hunt T.Biosci.Rep.,2002,22:465-486
[5]Li X,Lu F,Tian Q,et al.J.Neural Transm.,2005,113:93-102
[6]Vougolkov A,Dbilladeau D.Future Oncol.,2006,2:91-100
[7]Mondragon-Rodriguez S,Perry G,Zhu X,et al.Int.J. Alzheimer′s Dis.,2012,2012:1-4
[8]Rix L L,Kuenzi B M,Luo Y,et al.ACS Chem.Biol.,2014,9: 353-358
[9]Fang X,Yu S X,Lu Y,et al.PNAS,2000,97:11960-11965
[10]Goedert M,Spillantini M G,Crowther R A.Brain Pathol., 1991,1:279-286
[11]Leroy K,Boutajangout A,Authelet M,et al.Acta Neuropathol., 2002,103:91-99
[12]Bradley C A,Peineau S,Taghibiglou C,et al.Front.Mol. Neurosci.,2012,5:1-11
[13]Rudenko A,Seo J,Hu J,et al.J.Neurosci.,2015,35:2372-2383
[14]Engmann O,Giese K P.Front.Mol.Neurosci.,2009,2:1-5
[15]Leclerc S,Garnier M,Hoessel R,et al.J.Biol.Chem.,2001, 276:251-260
[16]Mettey Y,Gompel M,Thomas V,et al.J.Med.Chem.,2003, 46:222-236
[17]Tnguyen T,Jtepe J.Curr.Med.Chem.,2009,16:3122-3143
[18]Crunkhorn S.Nat.Rev.Drug Discovery,2015,14:457-457
[19]Berg S,Bergh M,Hellberg S,et al.J.Med.Chem.,2012,55: 9107-9119
[20]Chen Q,Cui W,Cheng Y,et al.J.Mol.Model.,2011,17: 795-803
[21]Boulahjar R,Ouach A,Matteo C,et al.J.Med.Chem.,2012, 55:9589-9606
[22]Ouach A,Boulahjar R,Vala C,et al.Eur.J.Med.Chem., 2016,115:311-325
[23]Dessalew N,Bharatam P V.Eur.J.Med.Chem.,2007,42: 1014-1027
[24]Larkin M,Blackshields G,Brown N P,et al.Bioinformatics, 2007,23:2947-2948
[25]Case D A,Cheatham T A,Simmerling C L.AMBER 10, University of California.San Francisco:San Francisco,CA, 2008.
[26]Bayly C I,Cieplak P,Cornell W D,et al.J.Phys.Chem., 1993,97:10269-10280
[27]Cieplak P,Cornell W D,Bayly C,et al.J.Comput.Chem., 1995,16:1357-1377
[28]Fox T,Kollman P A.J.Phys.Chem.B,1998,102:8070-8079
[29]Mapelli M,Massimilinao L,Crovace C.J.Med.Chem., 2005,48:671-679
[30]Shin D,Lee S C,Heo Y S,et al.Bioorg.Med.Chem.Lett., 2007,17:5686-5689
[31]Zhao P,Li Y,Gao G,et al.Eur.J.Med.Chem.,2014,86: 165-174
[31]Wu Q,Kang H,Tian C,et al.Mol.Inf.,2013,32:251-260
[33]Aixiao L,Florent B,Franois M,et al.J.Mol.Struct. THEOCHEM,2008,849:62-75
[34]Wang J,Wolf R M,Caldwell J W,et al.J.Comput.Chem., 2004,25:1157-1174
[35]Cornell W D,Cieplak P,Bayly C I,et al.J.Am.Chem. Soc.,1995,117:5179-519
[36]Shiekhattar R,Mermelstein F H,Fisher R P,et al.Nature, 1995,374:283-287
[37]Ryckaert J,Ciccotti G,Cberendsen H.J.Comput.Phys., 1977,23:327-341
[38]Darden T,Myork D,Gpedersen L.J.Chem.Phys.,1993,98: 10089-10092
[39]Hou T,Wang J,Li Y,et al.J.Chem.Inf.Model.,2011,51: 69-82
[40]Andricioaei I,Karplus M.J.Chem.Phys.,2001,115:6289-6292
[41]Gu Y,Wang W,Zhu X,et al.J.Mol.Model.,2014,20:1-12 [42]Andricioaei I,Karplus M.J.Chem.Phys.,2001,115:6289-6292
[43]Wang W,Cao X,Zhu X,et al.J.Mol.Model.,2013,19: 2635-2645
[44]Sa R,Fang L,Huang M,et al.J.Phys.Chem.A,2014,118: 9113-9119
[45]Li C,Ma N,Wang Y,et al.J.Phys.Chem.B,2014,118: 1273-1287
[46]Ichiye T,Karplus M.Proteins,1991,11:205-217
[47]Schrodinger LLC.PyMOL Molecular Graphics System, Version 1.8,2015.
[48]Pradeep H,Rajanikant G K.Mol.Diversity,2012,16:553-562
[49]Dajani R,Fraser E,Roe S M,et al.Cell,2001,105:721-732
[50]Dajani R,Fraser E,Roe S M.EMBO J.,2003,22:494-501
[51]Lobanov M Y,Bogatyreva N S,Galzitskaya O V.Mol.Biol., 2008,42:623-628
[52]ZHANG Chuan(张川),ZHANG Lu-Jia(张鲁嘉),ZHANG Yang(张洋),et al.Acta Chim.Sinica(化学学报),2016,74: 74-80
[53]Otyepka M,Bartova I,Kriz Z,et al.J.Biol.Chem.,2006, 281:7271-7281
[54]Safi M,Lilien R H.J.Chem.Inf.Model.,2012,52:1529-1541
[55]Zhan D,Yu L,Jin H,et al.Int.J.Mol.Sci.,2014,15:17284-17303
[56]Laskowski R A,Swindells M B.J.Chem.Inf.Model.,2011, 51:2778-2786
Exploring the Selectivity of Tetrahydropyrido[1,2-a]isoindolone Derivatives to GSK3β and CDK5 by Computational Methods
DONG Ke-Ke YANG Xue-Yu ZHAO Teng-Teng ZHU Xiao-Lei*
(State Key Laboratory of Materials-Oriented Chemical Engineering,College of Chemical Engineering,Nanjing Tech University,Nanjing 210009,China)
Tetrahydropyrido[1,2-a]isoindolone derivatives are potent inhibitors of glycogen synthase kinase 3β (GSK3β)instead of homologous cyclin-dependent kinase 5(CDK5).Molecular docking,molecular dynamics simulation,and MM/PBSA energy calculation are utilized to reveal the kinase inhibitors′selective mechanism at the molecular level for improving selectivity.Dynamic cross-correlation map(DCCM)analysis is applied to study the effect of the inhibitor on the interactions between each residue in CDK5 and GSK3β.The results of molecular docking indicate that the binding modes of three inhibitors with two kinases are especially similar,and residues in the binding pockets of two kinases are aligned with each other based on the sequence comparing analysis of crystal structures.The analysis of Root Mean Square Deviation(RMSD)with little fluctuation underlies the stability and reliability of systems.Its values of CDK5(~0.15 nm)are less than GSK3β(~0.17 nm),and the inhibitor with higher value holds stronger flexibility and conformational changes of kinases.In terms of energies,the electrostatic and van der Walls energies are the major interactions for differentiating the activity between thesame inhibitor and two kinases.And the polar solvation energy plays pivotal role in discriminating the selectivity of kinase inhibitor.The residue decomposition indicates that the residues Glu97 and Thr138 of GSK3β are the key residues for differentiating the inhibitor selectivity.On the other hand,in the aspect of inter-residue interaction in one kinase,results indicate that the dynamic correlation of residues is different during the binding process of CDK5 and GSK3β with inhibitors.The correlation of Thr138 in the hinge domain of GSK3β with that of residues Val135~Gln206 is positive,while the correlation of Gln85 and Cys83~Ala150 in CDK5 is unclear,which is a key factor to distinguish inhibitor selectivity.
tetrahydropyrido[1,2-a]isoindolone derivatives;molecular dynamics simulation;selectivity;stability
中国分类号:O643.1A
1001-4861(2016)11-1919-12
10.11862/CJIC.2016.263
2016-05-11。收修改稿日期:2016-09-12。
国家自然科学基金(No.20706029,20876073,91434109)资助项目。*通信联系人。E-mail:xlzhu@njtech.edu.cn
