分子动力学模拟别构抑制剂Efavirenz对HIV-1逆转录酶的作用
2016-11-08孟现美张少龙张庆刚山东师范大学物理与电子科学学院济南250014
孟现美 张少龙 张庆刚(山东师范大学物理与电子科学学院,济南250014)
分子动力学模拟别构抑制剂Efavirenz对HIV-1逆转录酶的作用
孟现美张少龙*张庆刚*
(山东师范大学物理与电子科学学院,济南250014)
为了理解非核苷类逆转录酶抑制剂(NNRTIs)与HIV-1逆转录酶(RT)的相互作用机制,利用新力场ff12SB对未结合和结合Efavirenz(EFV)逆转录酶的三种RT大分子体系分别进行了100 ns的长时间动力学模拟。通过分析EFV对RT结构的影响、不同残基柔性和不同体系构象的动力学行为等,发现EFV的结合会导致RT结构变化,从而影响RT的活性;证实了EFV的“分子楔”作用;还发现EFV的结合不但引起“拇指关节炎”,而且引起轻度“手指关节炎”;整个模拟过程中没有出现不同构象间的跃迁,但是无别构分子时的RT张开构象表现出明显的闭合倾向。这些结果有助于理解NNRTIs的抑制机制和RT构象变化的动力学性质。另外,还比较分析了模拟方法对计算结果的影响,对大分子体系的动力学模拟具有重要借鉴意义。
HIV-I逆转录酶;逆转录酶抑制剂;别构抑制剂;分子动力学模拟;构象
doi:10.3866/PKU.WHXB201511302
1 引言
逆转录酶(RT)在I型人体免疫缺陷病毒(HIV-1)复制过程中,可以催化病毒RNA基因组使之逆转录为双链DNA病毒前体,因此是发明抗艾滋病(anti-AIDS)新药的一个重要靶酶1。目前,以RT为靶治疗艾滋病的药物中,非核苷类RT抑制剂(NNRTIs)被认为是一类具有前景的特效药2-4。NNRTIs也称为别构抑制剂,结合在RT的非活性位,可以避免活性位点或底物的选择性压力,具有广谱抗HIV-1耐药性的特点。如图1A所示,RT是一个由p66和p51两个亚基组成的异二聚体5。p66亚基由DNA聚合酶和RNase H结构域构成,其中,DNA聚合酶结构域形状类似于握杯的右手形,它的3个子域形象地称之为手指(fingers)、手掌(palm)和拇指(thumb)子域,并通过所谓的连接子(connection)与RNase H结构域连接在一起。p51亚基具有与p66聚合酶结构域同样的氨基酸序列,但折叠方式不同。如图1B所示,在p66的拇指和手掌连接处有一个疏水性口袋结构(NNRTI-BP),NNRTIs可以进入并束缚在袋内6。图中显示的EFV (Efavirenz)属于第二代NNRTIs类药物。
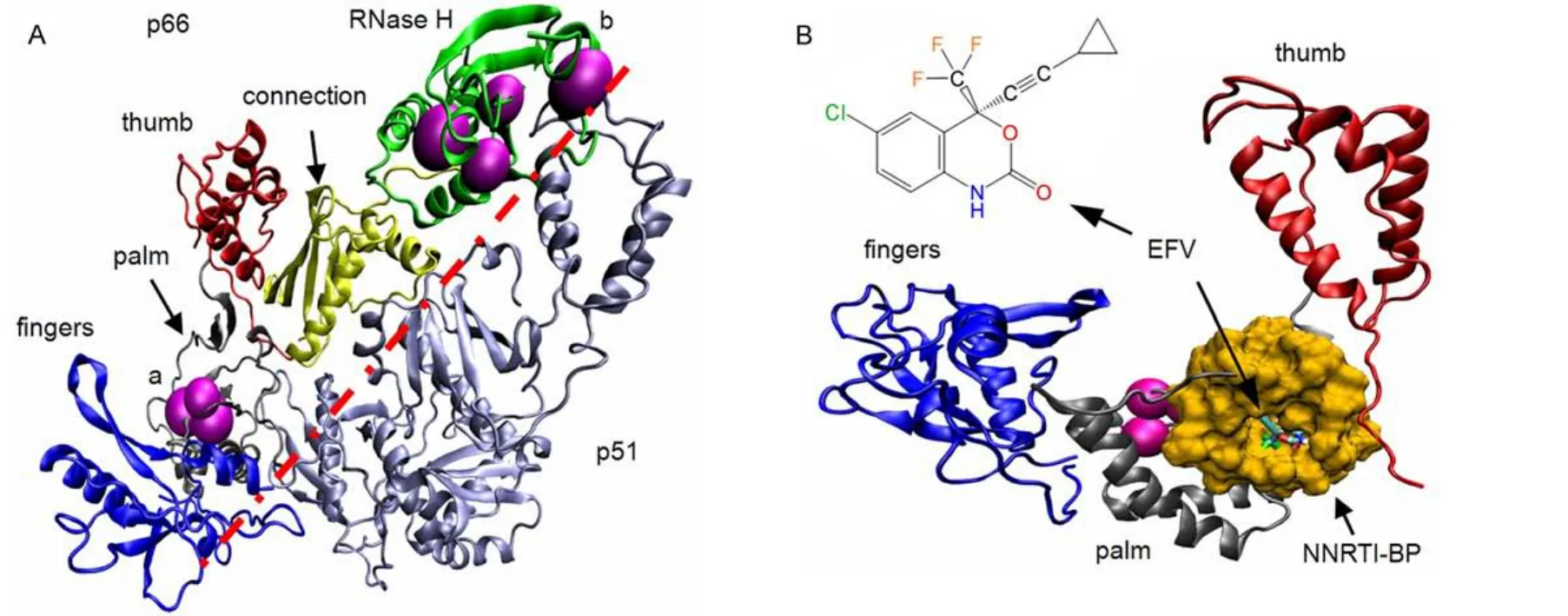
图1 逆转录酶(RT)和别构抑制剂(EFV)的结构Fig.1 Structures of reverse-transcriptase(RT)and the allosteric inhibitor(EFV)
然而,关于NNRTIs和RT的作用机制当前还无一致的理解。一般观点是建立在实验晶体数据分析基础上的“拇指关节炎”或“分子楔”模型7-9,但最近的荧光实验却意味NNRTIs的作用会使手指-拇指“握度松动(grip loosening)”,从而抑制聚合催化过程10。理论上也相应地开展了许多工作11-16,主要是利用分子动力学方法研究具有不同初始结构和不同底物的RT动力学性质。由于RT体系过大,所以模拟时间一般只有几个纳秒,所得计算结果也不同。例如,文献11(3 ns)和12(2.5 ns)的模拟结果支持“拇指关节炎”模型;文献13 (1 ns)模拟结果表明,去掉NNRTIs后拇指子域会迅速从张开塌向闭合状态转变;文献14(1.1 ns)模拟结果表明,底物会增加RT的柔性。近来,两个较长时间的动力学模拟结果更是引人关注:Ivetac和McCammon15利用GROMACS程序和GROMOS力场,对具有不同原子初速度的体系副本进行了30 ns的模拟,发现无论APO(无任何抑制剂)还是结合NNRTI的RT体系,其构象发生转变的几率都相当可观(分别为50%和25%);Wright等16用AMBER9和ff03力场进行的计算(模拟时间达100 ns,但步长较大为4 fs)发现,被别构抑制剂撑开的拇指子域可以重返类似APO的闭合状态。显然,这些模拟结果有悖于一般的“拇指关节炎”观点,意味着RT的构象变化和NNRTI的作用机制可能并不像一般认为的那样。换句话说,无论APO还是加上NNRTI后,RT体系的能量曲面上都可能存在两个极值点,分别对应张合状态,且两个极值点间过渡态的势垒不高,常温热力学运动就会引起从开放状态到闭合状态的转变。如果情况果真如此,药就将失去抑制功能,抗逆转录病毒治疗必须考虑这种情况。因此,应该弄清这些模拟结果的可靠性,或者说了解这些现象的发生几率。对这些问题的阐明将有助于深入理解NNRTIs的抑制机制和聚合酶结构域构象变化的动力学性质,从而为设计更有效的抑制剂提供理论启发。
分子动力学模拟结果的可靠性依赖于模拟方法和策略,先进的力场、较小的步长、较长的模拟时间以及体系动力学过程的充分稳定平衡将有助于改进计算结果,得到更可信的信息17-19。新的GPU大规模并行计算技术的出现为生物大分子模拟计算提供了更加高效的平台;新力场ff12SB20,21考虑到了侧链χ1扭转势的重要性仅次于骨架扭转势,不只对蛋白的主链φ′/ψ′扭转势参数做了进一步调整,并且也修正了17个氨基酸侧链的扭转势参数,从而改善了势函数的精度。
本文采用先进的ff12SB力场,使用GPU计算技术,利用AMBER12程序包20,对结合和未结合别构抑制剂EFV的三种RT体系进行了模拟计算。为了叙述简洁,结合EFV的RT体系简称EBR;未结合EFV,拇指子域为打开的RT体系简称为APOO,拇指子域为闭合的简称为APOC。研究目的是,(1)别构抑制剂EFV是如何结合到RT的非活性位并影响RT构象;(2)EFV结合到非活性位后对RT稳定性(即结构柔性)会产生怎样的影响;(3)通过先进的力场、较小的时间步长、较长的模拟时间和严格的模拟初始结构,重点探究不同初始构象条件下,RT拇指子域构象的动力学演化性质,以期验证张合两个状态间可能的转变信息,进而弄清NNRTIs和RT的作用机制。
2 理论计算方法
体系的初始构象取自蛋白质库(PDB ID:1DLO,1IKW和1HQU)。1DLO中只包含RT,1IKW中包含RT和EFV,1HQU中包含RT和HBY (别构抑制剂)。其中,所有带别构抑制剂的RT晶体结构都是不完整的,在1IKW中,p51亚基缺失残基217-231。由于1HQU的晶体结构具有较高的分辨率(0.27 nm),并结合了别构抑制剂,所以我们借助该晶体补全1IKW缺失的残基,补全的1IKW中,RT共拥有983个残基。另外,相对于1DLO,1IKW中的RT残基序列有两处发生变异,即p66亚基上S280C和E478Q。对于1DLO和1IKW中的RT,因为组成RT的各氨基酸参数预存在库文件中,所以直接利用AMBERTools12的LEaP模块,根据ff12SB力场为RT赋键参数和静电参数,并补全缺失的氢原子;对1IKW中的EFV,其力参数和静电信息是没有预存的,需要计算生成,我们利用sqm模块中的AM1-BCC拟合EFV的电荷分布,其力场参数采用GAFF力场参数22和拟合电荷时在Antechamber程序中生成的参数。这样,处理过的1IKW中的RT和EFV结合在一起组成体系EBR,RT单独作为体系APOO;处理过的1DLO中的RT作为体系APOC。三个体系外围都加了1 nm厚的截角八面体水盒子,并分别添加10、10和8个氯离子使之呈电中性。
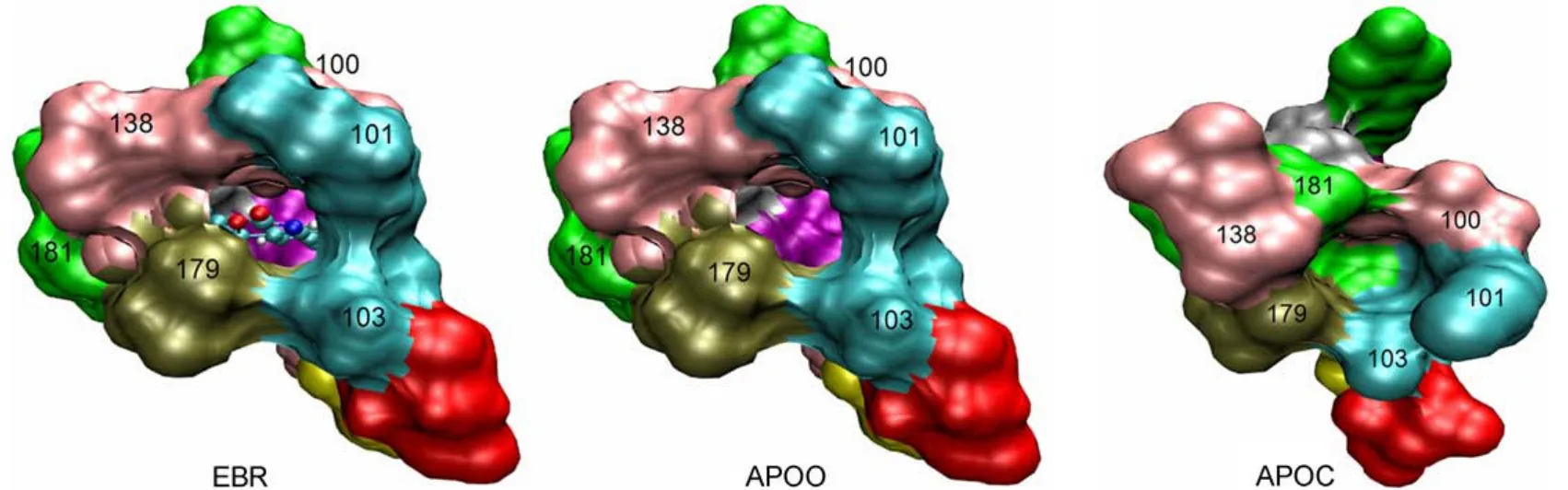
图2 三个NNRTI-BP体系中结合口袋入口处的初始构象Fig.2 Starting conformations of entrance of the binding pocket in the three RT systems
为了消除原子间的过近接触,采用AMBER12的PMEMD模块对三个体系进行能量最小化,先进行25000步最陡下降法计算,后进行25000步共轭梯度法计算,这样就得到了具有不同初始构象的三个模拟计算样本。图2示出了它们的别构袋口处构象,可以清楚看出,EBR、APOO和APOC的袋口分别处于张开、张开、闭合。接下来将上述三个体系的温度在300 ps内从0 K加热到300 K,然后进行200 ps的常温常压动力学平衡计算,最后对三个体系分别进行100 ns的分子动力学模拟。计算中,时间步长取2 fs,每隔5 ps记录一次坐标文件,并使用SHAKE算法限制含氢原子的键的变化23。模拟过程中同时计算监测系统的总能量、动能和密度随时间的变化情况。文中所有的结构图均采用VMD软件24,25完成。
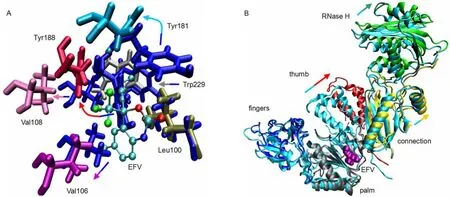
图3EFV与RT结合后引起的RT结构变化Fig.3 RT structure changes caused by binding EFV
3 结果与讨论
3.1EFV与RT结合引起的结构变化
NNRTI-BP由p66亚基上许多含芳环的残基(Tyr181、Tyr188、Phe227、Trp229、Tyr232),疏水残基(Lys101、Lys103、Ser105、Val106、Val108、Asp192、Glu224等)以及p51亚基的Glu138组成。口袋的入口位于p66和p51亚基的接合部,由p66亚基的Leu100、Lys101、Lys103、Val179、Tyr181和p51亚基的Glu138环绕组成(图2)7,26。通过图2可以看出,没有结合EFV的APOC体系,在NNRTIBP相应位置存在一个表面凹陷,袋口是闭合的。当EFV结合到RT上时,袋口张开,即RT结构发生了变化,这种变化可以分为结构上的短程和长程变化(图3)。短程变化如图3A所示,主要是构成NNRTI-BP的氨基酸位置和取向的变化。变化比较明显的是Tyr181(浅蓝色)、Tyr188(大红色)和Trp229(灰色)侧链,未结合EFV时,这三个氨基酸的芳环侧链指向疏水区核心,EFV的结合造成了侧链显著的扭转,使之远离疏水核心,从而制造出一个可以容纳小分子抑制剂的疏水空间。在这个过程中Val106(粉色)和Val108(紫红色)两个残基发生了明显的外向位移。进入疏水口袋的EFV通过氢键与芳环之间的π键和van der Waals等作用与口袋周围的氨基酸形成稳定的复合物。为了研究EFV导致的RT长程结构变化,将两个体系的手指子域中Cα原子进行叠合,如图3B所示,即保持手指位置相对不变,然后测量其它子域方位的相对变化。发现,EFV的结合使拇指子域外旋约38°,连接子和RNase H结构域的位置也分别远离手指12°和15°。另外我们还发现,RT结合EFV后,p51亚基上的手指、手掌、拇指子域和连接子也分别移动9°、12°、14°和18°。
3.2RMSD变化
均方根偏差(RMSD)是衡量体系稳定性的重要参量20,27,它反映蛋白质分子在动力学模拟过程中偏离初始结构的程度。我们使用AMBER12中的ptraj程序分析了EBR、APOO和APOC三体系的骨架碳原子Cα相对于初始结构的RMSD随时间的变化(图4)。可以看出,在整个模拟过程中,体系EBR(红线)结构的RMSD无论其数值大小还是涨落幅度均明显低于APOO(黑线)和APOC(蓝线),说明体系EBR的稳定性优于APOO和APOC。
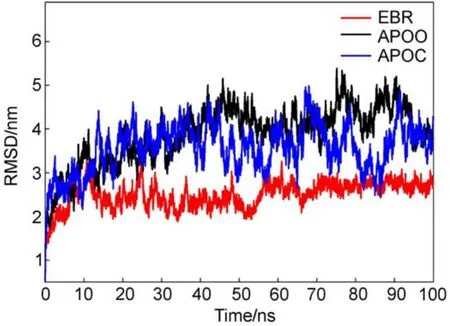
图4 三体系骨架碳原子Cα的RMSD随时间的变化Fig.4 Time dependences of root mean square deviation (RMSD)of the backbone Cαatoms for the three systems
具体看,EBR体系的RMSD变化情况比较简单,最初0-12 ns不太稳定,是一个由小变大趋于平衡的过程,12 ns之后直到模拟结束的100 ns,其值基本稳定在0.24 nm附近,上下浮动范围约为0.04 nm。对于APOO和APOC两个体系,在整个模拟时间内,其RMSD表现出了相似的特征,在开始的0-40 ns内,两个体系的RMSD无论在变化趋势还是波动幅度上几乎都是重合的,RMSD值由0.2 nm上升到0.4 nm;在之后的整个模拟时间内,APOO和APOC的RMSD也表现了相似的波动性,只是APOC的RMSD值较小波动幅度较大,二者的RMSD平均值分别约为0.40和0.36 nm,浮动范围约为0.04和0.10 nm。
EBR体系较小的RMSD值表明,结合别构抑制剂EFV后,RT结构稳定性变好,或者说EFV增加了体系的刚性,这正是一般认为的别构抑制剂NNRTIs的“楔子”作用,它阻止了相关子域的柔性。对于APOO和APOC两个体系,其RMSD在整个模拟时间内表现出的相似性意味着,没有抑制剂时,张开状态(APOO)与闭合状态(APOC)具有类似的动力学行为。值得注意的是,这时APOC的RMSD虽然具有较小的平均值但却具有很宽的波动幅度,其最大值与APOO的相近,而最小值却与EBR的相当,意味着APOC的状态很不稳定,结构变化忽大忽小。而与之相对,EBR总是维持在结构变化较小的状态,APOO倾向于保持在结构变化较大的状态。这说明APOO和APOC两体系比EBR具有较大结构柔性,而APOC的结构在动力学过程中更易发生变化,即时柔时刚。显然,不同体系所表现的这些不同动力学行为只有通过较长的模拟时间才能获得充分取样,这解释了为什么之前文献13,14通过短时间模拟得到的结果是矛盾的。
3.3B因子分析
B因子(B-factor)描述体系中各个残基在动力学过程中偏离其平均结构的程度,反映残基的柔软程度,可以用来研究分子动力学过程中蛋白的一些性质变化20。图5给出了EBR、APOO和APOC三体系中所有残基的B因子,它们取自模拟过程中体系的RMSD数值较小且稳定的时段,分别为,EBR:74-88 ns,APOO:12-32 ns,APOC:48-62 ns。
图5A是三体系中RT全部983个残基的B因子变化情况。可以看出,p51和p66亚基中距别构抑制剂EFV结合位较远的连接子和RNase H结构域,即320号之后的残基,三体系相应残基的B因子变化不大,说明别构抑制剂的加入对这些部位影响不大。然而,距离别构位较近的聚合酶活性区,包括手指、手掌和拇指子域,即1到320号残基,三体系相应残基的B因子差别非常明显。
拇指和手指子域的构象变化和柔性直接关系到RT的催化功能,所以有必要详细了解别构抑制剂对这两个子域构象的影响和作用机制,为便于观察,图5B放大了这些区域。可以看出,对于手指子域,APOC(蓝线)的B因子普遍较小,APOO(黑线)的普遍较大;EBR(红线)大部分与APOC相当甚至偏小,只有小部分(60-70,135-145)较大,与APOO相当。这说明无EFV结合时,闭合状态(APOC)下手指子域的柔性明显较小,而张开状态(APOO)下手指子域总体上表现出较大柔性。对于结合了EFV的体系EBR,其拇指子域的B因子普遍变小,表明别构抑制剂对拇指子域的柔性具有明显抑制作用,这正是所谓“拇指关节炎”效应;值得注意的是,这时手指子域大部分残基(1-60,70-86)的柔性也变小了,说明EFV对这些残基也具有抑制作用,手指也患了“关节炎”,只是症状较轻。对于手掌子域,三个体系的B因子差别不大,说明手掌子域具有较强的刚性,手指拇指的张合以及EFV的加入对手掌子域骨架支撑结构的影响不大。
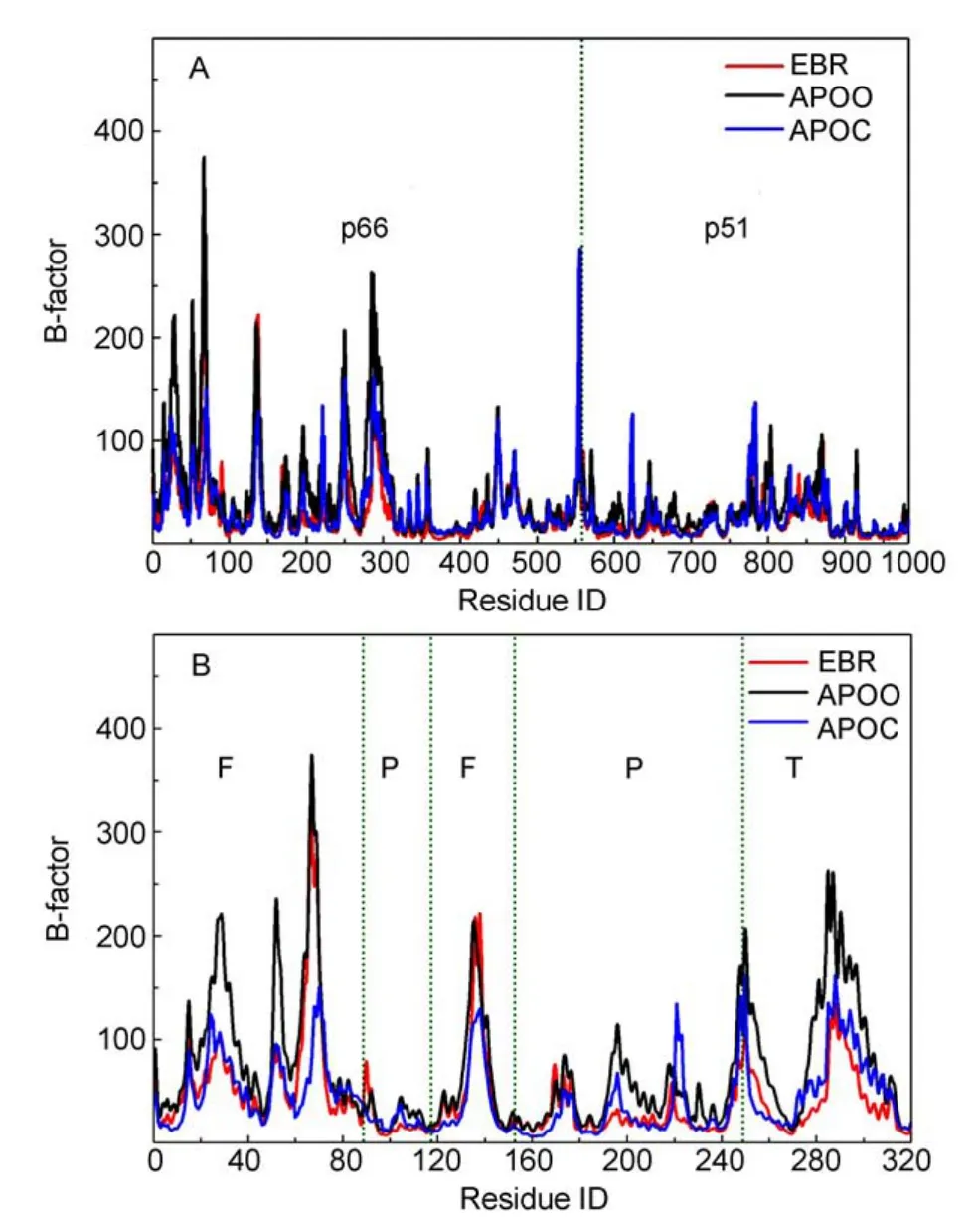
图5 三个RT体系全部残基(A)和聚合酶活性区残基(B)的B因子Fig.5 B-factors of all the residues of the three RT systems(A)and the polymerase domain(B)
综上所述,别构抑制剂EFV对手掌子域骨架残基的刚性影响很小;对拇指子域绝大部分残基的柔性有明显抑制;对RT手指子域大部分残基的柔性也有抑制作用。这个结果有利地证明了关节炎模型。而且,我们的结果表明EFV不但引起“拇指关节炎”,也使手指患上了轻度“关节炎”,这是之前文献没有报道的。
3.4构象变化
蛋白分子中某些原子间的距离变化常常能反映整个体系构象的变化,这种距离分析方法可以给出直观清楚的构象变化信息。为了理解RT上手形构象的动力学性质,我们计算了手指子域顶端Lys66和拇指子域顶端Leu289的Cα原子之间的距离L(图6A)在动力学过程中的变化。这个距离由于是拇指和手指远端间的距离,因此能够较好地反映手形的张合程度。
图6B直观地给出了动力学过程中三个体系手形区域平均结构的叠合示意图。三个平均结构均取自RMSD值较小且稳定的模拟时段,分别为,EBR:74-88 ns,红色;APOO:12-32 ns,黄色;APOC:48-62 ns,蓝色。可以看出,三个体系的拇指子域结构表现出明显差异:APOC的拇指子域向手指子域方向大幅度靠拢,发生握合;相对APOO,EBR的拇指子域发生了明显的向外张开偏移。然而比较起来,三体系手指子域的构象变化很小。这说明别构抑制剂或RT初结构对体系动力学结构的影响主要体现在拇指子域,或者说拇指子域的构象对RT的性质(结构及由此引起的活性)起着重要作用,这个直观图像与实验上的“拇指关节炎模型”相符。
图6C清楚地表明,无“楔子”的闭合状态下(APOC),L在整个动力学过程中基本不变,保持在1.49 nm附近;EBR和APOO两个体系的L值大小及波动幅度相似,其值都在3.8 nm左右,变化幅度约为±0.5 nm。这个结果给出的物理图像是:在整个动力学模拟过程中,APOC保持握紧的手形;EBR和APOO则保持张开的手形,而且二者张开的程度相当。图6D给出了三个体系中L值的几率(时间)分布函数P,它不但反映手形的张合程度,更重要的是它反映了系统构象的稳定程度。因为,一个确定的L值对应一个确定的构象或手形,所以P函数曲线的形状对应构象的变化程度或者结构稳定程度。图6D中,APOC的P(L)分布(蓝线)是一高耸的中心位于1.49 nm的单峰,表明该体系在整个动力学模拟过程中,处于一种稳定的构象结构,从自由能曲面上看,代表闭合状态对应一个较深的能量极小值点,体系的相点被牢牢的束缚在极值点附近做小幅振动。特别是,1.49 nm处的峰不但高耸尖锐而且对称性极高,这意味无别构分子加入时,握合的手形是一个非常稳定的状态,除非有外力作用,在等温平衡热力学条件下不会自动转变成张开状态。这就是说,正常情况下,RT总是倾向于闭合的活性状态,具有加工合成病毒的功能。这个结果与实验一致,但不同于Ivetac和McCammon15先前的模拟结果,他们通过改变体系中原子的初始速度使四个副本中的两个发生了由闭合到张开的状态跃迁。值得注意的是,这种构象变化都发生在模拟运行的初期,因此可以推断,这种跃迁与原子初速度的赋值有关,即这些发生跃迁的样本体系相点的初速度刚好处于通向势垒较低的势能峡谷中,从而在程序对体系增温(对原子速度乘以大于1的系数)至300 K时,推动体系越过势垒进入另一能量极值点。另外,计算误差和力场误差也都可能引起不准确的结果。EBR体系的P(L)也是一个对称性良好,中心位于3.84 nm处的单峰,表明体系也始终稳定在相应的自由能极值点附近,这正是所谓的“分子楔”作用。比较而言,APOO的P(L)是一个不对称分布,在主峰3.74 nm左侧相距约0.30-0.70 nm处出现了一台梯。从构象上说,这个台梯结构反映了体系的构象由张开向闭合转变的趋势或征兆。换言之,尽管APOO大部分时间处于张开的初构象,但与EBR不同的是,它存在由张开向闭合转变的明显倾向,并有相当的几率处于半握合状态,这个结果也与实验一致,即自然的APOO总是趋于具有活性的闭合构象。
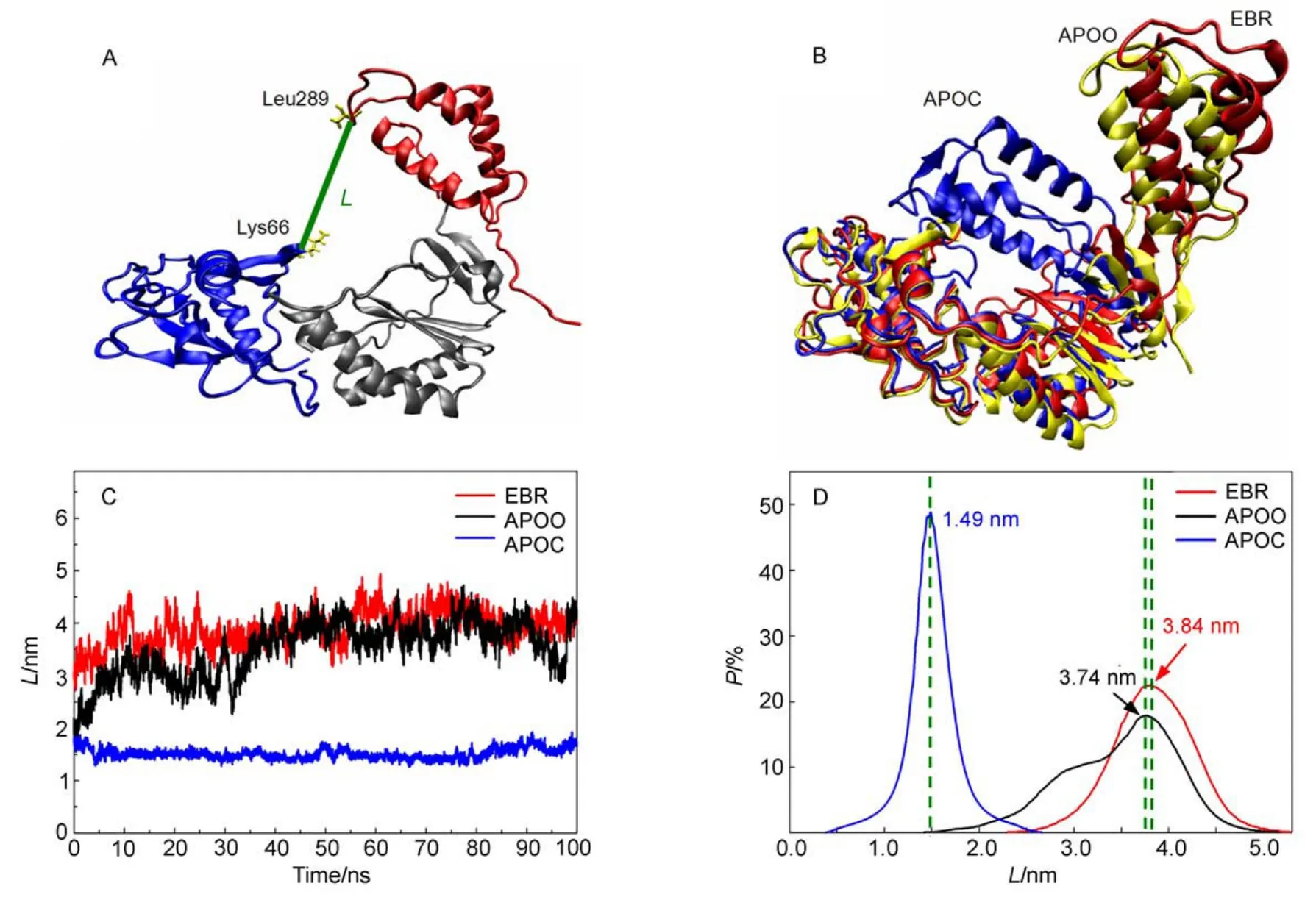
图6 100 ns动力学模拟过程中RT手形握合状态的变化Fig.6 Conformation changes of the RT hand opening and closing in the 100 ns dynamics simulations
综上所述,可以看出,相对于RMSD和B因子分析,手指顶端Lys66和拇指顶端Leu289的Cα原子间距离L的分析给出了体系在动力学过程中更加直观的物理图像和更加清晰的动力学性质。我们的结果支持基于实验晶体资料基础上的“拇指关节炎”或“分子楔”模型,并与文献11,12报道的模拟结果一致。第一次揭示了APOO由张开状态向闭合构象转变的倾向和动力学性质,即APOO的张开构象也是一个相对稳定的能量极点,只是相应于闭合方向的势能面比较低平,这成为APOO由张向合转变的驱动力。我们没有得到文献16报道的构象跃迁现象,原因可能是所用力场不同,也可能是他们使用较大时间步长积累的误差所致,更可能与模拟开始时原子的初速度随机赋值及随后的加温过程有关,因为文献报道的构型转变均发生在模拟开始阶段。
4 结论
运用GPU大规模并行计算技术,采用AMBER12和ff12SB力场,对三种RT大分子体系分别进行了100 ns较长时间的分子动力学模拟,研究了别构抑制剂的结合对体系结构和不同子域残基柔性的影响。主要结论是:(1)当别构抑制剂EFV结合到RT上的特定别构位后,会导致体系结构的变化,这个变化会影响RT的活性,从而实现别构抑制剂对RT功能的控制。(2)通过系统地分析相关体系的RMSD以及残基的B因子,发现对于RT,别构抑制剂的作用不但撑开拇指和手指子域,而且增强了两个子域残基的刚性,证实了“分子楔”模型观点。值得说明的是我们的结果不但验证了“拇指关节炎”观点,而且预测了轻度“手指关节炎”症状。(3)通过手指顶端Lys66和拇指顶端Leu289的Cα原子间距离L的分析发现,无别构抑制剂加入时,握合的手形是一个非常稳定的状态,即自然条件下,RT总是倾向于闭合的活性状态,具有加工合成病毒的功能。EBR体系的张开状态也是一个稳定状态。比较而言,尽管APOO大部分时间处于张开的初构象,但与EBR不同的是,它存在由张开向闭合转变的明显倾向,并有相当的几率处于半握合状态,说明自然的APOO倾向于具有活性的闭合构象。这些结果与实验一致,并证实了别构抑制剂NNRTIs的“分子楔”作用。另外,通过比较不同文献报告的模拟结果,我们认为,动力学模拟结果可能对模拟方法和策略具有很强的依赖性,包括模拟时间长短、时间步长的大小、模拟开始结构的准备、力场的精度以及原子初速度的随机赋值(原子的初速度应具有热力学平衡分布的特点,即Maxwell分布)等,精确的力场和模拟过程中体系的良好动力学平衡是得到可靠模拟结论的必要条件。最后强调的是,我们的计算结果表明,一旦RT体系稳定在开放或闭合状态,则状态改变是一种小概率事件,常温下(300 K)仅靠热力学效应似乎很难使体系在两态之间发生跃迁。对这些问题的阐述将有助于更加详细地理解NNRTIs的抑制机制和聚合酶结构域构象变化的动力学性质,从而为设计更有效的抑制剂提供理论依据。然而,要得到这些问题的确切结论,需要进行多样本和更长时间的分子动力学模拟计算。希望我们的工作能给其他研究者,尤其是从事模拟计算的人员带来启发。
References
(1)Mathers,C.D.;Loncar,D.PLoS Med.2006,3(11),e442. (2)De Clercq,E.Chem.Biodivers.2004,1,44.
(3)Ren,J.;Stammers,D.K.Trends Pharmacol.Sci.2005,26, 4.doi:10.1016/j.tips.2004.11.003
(4)Zhu,R.X.;Wang,F.;Liu,Q.;Kang,T.G.Acta Chim.Sin. 2011,69(15),1731.[朱瑞新,王飞,刘琦,康廷国,化学学报,2011,69(15),1731.]
(5)Jacobo-Molina,A.;Arnold,E.Biochemistry 1991,30(26), 6351.doi:10.1021/bi00240a001
(6)Lawtrakul,L.;Beyer,A.;Hannongbua,S.;Wolschann,P. Monatsh.Chem.2004,135(8),1033.
(7)Sluis-Cremer,N.;Temiz,N.A.;Bahar,I.Curr.HIV Res.2004, 2(4),323.doi:10.2174/1570162043351093
(8)Bakan,A.;Bahar,I.Proc.Natl.Acad.Sci.U.S.A.2009,106 (34),14349.doi:10.1073/pnas.0904214106
(9)Kohlstaedt,L.A.;Wang,J.;Friedman,J.M.;Rice,P.A.; Steitz,T.A.Science 1992,256(6),1783.doi:10.1126/ science.1377403
(10)Liu,S.X.;Abbondanzieri,E.A.;Rausch,J.W.;Le Grice,S.F. J.;Zhuang,X.W.Science 2008,322(5904),1092.doi: 10.1126/science.1163108
(11)Shen,L.L.;Shen,J.H.;Luo,X.M.;Cheng,F.;Xu,Y.C.; Chen,K.X.;Arnold,E.;Ding,J.P.;Jiang,H.L.Biophys.J. 2003,84(6),3547.doi:10.1016/S0006-3495(03)75088-7
(12)Zhou,Z.;Madrid,M.;Evanseck,J.D.J.Am.Chem.Soc.2005, 127(49),17253.doi:10.1021/ja053973d
(13)Madrid,M.;Jacobo-Molina,A.;Ding,J.;Arnold,E.Proteins 1999,35(3),332.
(14)Madrid,M.;Lukin,J.A.;Madura,J.D.;Ding,J.;Arnold,E. Proteins 2001,45(3),176.
(15)Ivetac,A.;McCammon,A.J.J.Mol.Biol.2009,388(3), 644.doi:10.1016/j.jmb.2009.03.037
(16)Wright,D.W.;Sadiq,S.K.;De Fabritiis,G.;Coveney,P.V. J.Am.Chem.Soc.2012,134(31),12885.doi:10.1021/ ja301565k
(17)Chen,J.Z.;Wang,J.N.;Zhu,W.L.;Li,G.H.J.Comput. Aided Mol.Des.2013,27(11),965.
(18)Luo,F.;Gao,J.;Cheng,Y.H.;Cui,W.;Ji,M.J.Acta Phys.-Chim.Sin.2012,28(9),2191.[罗芳,高剑,成元华,崔巍,计明娟.物理化学学报,2012,28(9),2191.] doi:10.3866/PKU.WHXB201207063
(19)Zhang,H.;Lu,J.R.;Mu,J.B.;Liu,J.B.;Yang,X.Y.;Wang, M.J.;Zhang,R.B.Acta Phys.-Chim.Sin.2015,31(10),566. [张贺,卢俊瑞,穆江蓓,刘金彪,杨旭云,王美君,张瑞波,物理化学学报,2015,31(10),566.]doi:10.3866/PKU. WHXB201501061
(20)Case,D.A.;Darden,T.A.;Cheatham,T.E.,III;Simmerling, C.L.;Wang,J.;Duke,R.E.;Luo,R.;Walker,R.C.;Zhang, W.;Merz,K.M.;Roberts,B.P.;Hayik,S.;Roitberg,A.E.; Seabra,G.;Swails,J.M.;Kolossváry,I.;Wong,K.F.;Paesani, F.;Vanicek,J.;Wolf,R.M.;Liu,J.;Wu,X.;Brozell,S.R.; Steinbrecher,T.;Gohlke,H.;Cai,Q.;Ye,X.;Wang,J.;Hsieh, M.J.;Cui,G.;Roe,D.R.;Mathews,D.H.;Seetin,M.G.; Salomon-Ferrer,R.;Sagui,C.;Babin,V.;Luchko,T.;Gusarov, S.;Kovalenko,A.;Kollman,P.A.AMBER 12;University of California:San Francisco,CA,2012.
(21)Meng,X.M.;Wang,J.L.;Zhang,S.L.;Zhang,Q.G.ActaChim.Sin.2013,71(8),1167.[孟现美,王加磊,张少龙,张庆刚,化学学报,2013,71(8),1167.]doi:10.6023/A13030327
(22)Lindorff,L.K.;Piana,S.;Palmo,K.;Maragakis,P.;Klepeis,J. L.;Dror,R.O.;Shaw,D.E.Protein Force Field 2010,78(8), 1950.
(23)Chen,J.Z.;Zhang,D.L.;Zhang,Y.X.;Li,G.H.Int.J.Mol. Sci.2012,13(2),2176.
(24)Humphrey,W.;Dalke,A.;Schulten,K.J.Mol.Graphics 1996, 14(1),33.doi:10.1016/0263-7855(96)00018-5
(25)The Theoretical and Computational Biophysics Group.VMD, Revision 1.8.7;NIH Center for Macromolecular Modeling and Bioinformatics,the Beckman Institute,University of Illinois at Urbana-Champaign.
(26)Beyer,A.;Lawtrakul,L.;Hannongbua,S.;Wolschann,P. Monatsh.Chem.2004,135(7),1047.
(27)De Clercq,E.Nat.Rev.Drug Discov.2007,6(12),1001. doi:10.1038/nrd2424
Effect of the Allosteric Inhibitor Efavirenz on HIV-1 Reverse Transcriptase by Molecular Dynamics Simulation
MENG Xian-MeiZHANG Shao-Long*ZHANG Qing-Gang*
(School of Physics and Electronics,Shandong Normal University,Jinan 250014,P.R.China)
To understand the allosteric modulation dynamics of non-nucleoside reverse transcriptase inhibitors (NNRTIs),various models and suggestions have been derived from crystallography and simulation.Here,using a new force field,ff12SB,and GPU parallel computing technology,we performed 100-ns-long molecular dynamics simulations on three reverse transcriptase(RT)systems,one bound to inhibitor Efavirenz(EFV)and the others free.Analyses of the influence of the EFV on the conformation of the RT,flexibility of residues and dynamic behaviors of the systems were conducted.The simulations indicate that EFV binding induces structural distortion of the RT,whereas the configuration of the RT is more stable during dynamics,along with a decreasing extent of motion of the residues.EFV suppresses the flexibility of the thumb subunit and reduces that of most residues in the fingers subdomain as well,suggesting that EFV causes not only the so-called“thumb arthritis”but also a slight“fingers arthritis”.No conformational transition occurred throughout the entire simulations and the samples maintained their starting conformations,i.e.,free RT with a closed conformation stayed in the functional state and EFV-bound RT remained in open conformation.However,EFV-free RT with an initially open conformation exhibited an evident trend toward the closed state.These results agree with the models from experiments,and present a useful insight into the allosteric inhibition mechanism of NNRTIs.In addition,the simulation methodology has been discussed in detail and will be of significance to the computational simulation of large biological molecules.
HIV-1 reverse transcriptase;Nonnucleoside reverse transcriptase inhibitor; Allosteric inhibitor;Molecular dynamics simulation;Conformation
September 14,2015;Revised:November 25,2015;Published on Web:November 30,2015.*Corresponding authors.ZHANG Qing-Gang,Email:zhangqg@sdnu.edu.cn;Tel:+86-531-86180349. ZHANG Shao-Long,Email:slzhang@sdnu.edu.cn;Tel:+86-531-86182521.
O641
The project was supported by the National Natural Science Foundation of China(11274206).
国家自然科学基金(11274206)资助项目
©Editorial office ofActa Physico-Chimica Sinica
