猪小体型相关受选择基因位点的研究
2016-11-08李完波朱亚玲艾华水郭添福
李完波,朱亚玲,艾华水,郭添福
(江西农业大学省部共建猪遗传改良与养殖技术国家重点实验室,南昌 330045)
猪小体型相关受选择基因位点的研究
李完波*#,朱亚玲#,艾华水,郭添福*
(江西农业大学省部共建猪遗传改良与养殖技术国家重点实验室,南昌 330045)
旨在通过对比大、小体型两组中国地方猪种在基因组上的遗传分化,检测出全基因组选择信号,以期能鉴别出影响猪体型大小的相关基因及可能的突变位点,解析小体型猪种形成的分子机制。本研究选用小体型的五指山猪和巴马香猪,以大体型的金华猪、二花脸、河套大耳猪为对照,基于全基因组重测序数据,使用两组猪群间的遗传分化系数(Fst)和小体型猪组内的杂合度(Heterozygosity) 检测体型选择信号。Fst值较大而杂合度小的染色体区段作为体型相关的受选择候选区域。对这些候选区域所包含的基因进行基因功能及通路的注释分析、以及突变位点的搜寻,试图揭示与猪体型大小相关的可能受选择的基因和突变。结果,通过筛选全基因组重测序数据,我们得到32 475 218个高质量的SNPs,利用40 kb的滑动窗口进行Fst和杂合度的计算。Fst值经Z转换(Z(Fst))后大于5的区段有242个。取杂合度最小值的前200个区间与242个Z(Fst)>5的区间的交集,确定了13个受选择候选区域,其中共包含28个基因。通过PANTHER进行基因注释,发现了17个与体型性状相关的候选基因,包括IGF1R、GUCY1A3等功能候选基因。本研究基于高通量测序数据,结合群体遗传学分析手段,确定了与体型大小相关的受选择区域,综合分析推测五指山和巴马香小体型猪的形成可能是由于处于非基因编码区域的一个或者多个突变造成。
小型猪;全基因组重测序;选择信号;体型大小;候选基因
猪是人类较早驯化的家养动物之一。由于受到较强的人工选择及对所处圈养环境的适应,家猪品种在体型外貌、生长性能和行为习惯等重要性状上与野猪有了明显分化。中国南方地区有着丰富的小型猪猪种资源,如五指山猪、巴马香猪、从江香猪、剑白香猪、环江香猪、滇南小耳猪等,其一般都具有体型小、生长缓慢、肉质细嫩和耐近交的特点[1]。小型猪的成年个体体重多在40 kg以下,而五指山猪成年个体体重更低于25 kg[2]。与小型猪相比,分布在中国南方和北方的大型猪的体型明显要大,成年猪体重一般超过100 kg[3]。而猪的体型大小和生长速度一直都是商业猪种选育的重点,在遗传上解析小型猪的形成机制,并定位相关的突变基因位点,对猪种遗传差异的探索及品种的选育都具有重要的指导意义。此外,猪在器官大小和生理特性等方面和人类接近,而小型猪体型小、饲养经济、便于管理的优点可作为人类医学模式动物的理想材料。在弄清小型猪遗传机制的基础上,通过常规或基因工程育种方法进一步缩小我国现存的小型猪猪种的体型,将有可能推动小型医学试验猪在更大范围的应用。
随着第二代高通量测序技术成本的大幅下降,全基因组重测序的策略被大量应用于遗传学和基因组学研究中。近年来,基于全基因组重测序数据,通过检测基因组范围内的选择信号(Signatures of selection),大量与驯化、适应性和重要性状相关的受选择区段被相继报道。其中,涉及的研究对象有鸡、牛、狗、猪、羊等[4-7]。对单一品种或群体进行选择信号检测时,很难确定该区段是真正的受选择区域还是由于遗传漂变造成的。本研究借鉴前人的策略,将五指山猪和巴马香猪合为小型猪组,将金华猪、二花脸猪和河套大耳猪合为大型猪组。同时,在大、小体型猪组间应用群体分化系数(Fst)初步确定两组间的受选择区域。Fst利用了群体间存在的较显著的等位基因频率差异来检测选择信号,但它并不能说明到底是哪个群体存在选择,即选择方向不明确。因此,结合小型猪群体的杂合度(Heterozygosity)分析,进一步确定小型猪中特异受选择的区段,并以Fst和杂合度分析结果的重叠区间做为小型猪中受选择的候选区间,从而可以较为准确的确定与体型大小相关的受选择区间。
本研究利用公共数据库和本试验新增的小型猪和大型猪的全基因组测序数据,通过群体遗传学方法检测全基因组选择信号,以期解析中国小型猪形成的分子机理,为猪种遗传改良及开发实用的小型医用模式猪提供理论基础。
1 材料与方法
1.1试验材料
本研究选用小型猪五指山猪6头、巴马香猪12头,大型猪金华猪6头、二花脸猪6头和河套大耳猪6头作为试验材料(表1)。五指山猪、巴马香猪、二花脸猪、河套大耳猪全基因组重测序数据从H.S.Ai等[8]发表的数据中获得。金华猪重测序数据是基于Illumina公司Hiseq 4000平台产生。
1.2全基因组重测序数据的处理
质控过滤后的测序reads通过BWA软件(http://bio-bwa.sourceforge.net/)比对到猪参考基因组susScr3版本(http://hgdownload.soe.ucsc.edu/goldenPath/susScr3/bigZips/)。 Picard软件(http://picard.sourceforge.net)用来去除重复reads。其后,使用GATK(The Genome Analysis Toolkit)软件[9],将比对后得到的bam文件在含有插入或缺失突变的区间进行区域内重新比对,并进行碱基测序质量校正(Recalibration)。通过使用GATK HaplotypeCaller算法,针对每一个测序个体生成一个gVCF文件。最后,使用所有试验个体的gVCF文件产生了单核苷酸多态位点(SNP)和短插入缺失(INDEL)数据集。通过使用GATK的SelectVariants功能选择出高质量SNP位点,并设定如下的SNP过滤标准“QD>2.0 && MQ>40.0 && FS<60.0 && ReadPosRankSum>-8.0 && MQRankSum>-12.5”,舍弃含3个以上等位基因的位点,最终获得了一套高质量SNP数据集用于群体遗传学分析。
表1试验猪样本信息表
Table 1The summary information for sampled pigs

分组Group品种Breed个体数Numberofanimals测序深度Depthofcoverage金华猪6~26×大体型猪Large-size二花脸猪6~21×河套大耳猪6~20×小体型猪Small-size五指山猪6~21×巴马香猪12~22×
1.3选择信号检测
本研究采用VCFtools (http://vcftools.sourceforge.net/),以40 kb染色体区间为滑动窗口,每次滑动20 kb来计算大、小体型猪之间的Fst。VCFtools采用C.C.Cockerham 和B.S.Weir于1984年发表的算法来计算Fst值[10]。同时,以相同大小的滑动窗口策略进行小型猪组内期望杂合度的计算。滑动窗口的期望杂合度为区间内每个SNP位点期望杂合度的平均值,该计算使用Python代码完成。因有些区间可能是调控序列受选择,为尽可能囊括靶向基因,在候选受选择区域上下游各加100 kb再继续下游分析。
1.4受选择区域的候选基因注释、GO分析和通路分析
由于猪参考序列基因注释不完善,猪中注释的RefSeq转录本仅有5 305个,相较于人和老鼠的基因数差距很大。为避免在后续分析中遗漏基因,通过利用UCSC Genome Browser (http://genome.ucsc.edu/)在受选择区间选出猪中已注释的RefSeq基因,同时,将人和小鼠中同时注释了的同源基因(猪中暂无注释的)也纳入后续分析。通过这种策略,大大扩充了后续分析基因集,有效避免了遗漏未注释的基因。其后,采用PANTHER (http://www.pantherdb.org/)和DAVID (https://david.ncifcrf.gov/)两个软件对候选基因的功能和通路进行注释分析。此外,还系统检索了猪的QTL数据库(http://www.animalgenome.org/cgi-bin/QTLdb/SS/index),分析与体重、体长和生长等性状相关的QTL是否在受选择区域富集。
1.5基因突变注释
为找到可能影响小型猪体型形成的关键突变,本研究对13个候选区域里的变异进行注释。通过应用本地版Variant Effect Predictor (VEP)(http://www.ensembl.org/info/docs/tools/vep/index.html)软件,对候选基因所包含的所有SNP和插入缺失突变进行注释。同时,对可能显著影响基因功能、结构的突变位点,还进行了多物种间的序列保守性分析。VEP软件运行代码:
perl variant_effect_predictor.pl --database --coding_only --sift b --species sus_scrofa --input_file Input_selected.vcf --force_overwrite。
2 结 果
2.1候选受选择区域
本试验利用常染色体40 kb滑动窗口计算区间Fst值。所得Fst平均值为0.092,方差为0.049,接近正态分布,将Fst进行标准化得到Z(Fst)。Z(Fst)越大表明群体间遗传分化程度越大。以相同滑动窗口策略计算五指山和巴马香猪群体的杂合度(Heterozygosity),得到区间杂合度平均值为0.342,方差为0.043,符合正态分布,标准化后得到Z(H)。通过Z(Fst)值可以看出,大、小体型猪种在很多染色体区间存在遗传分化,最显著的区间出现在8号染色体上(图1a,表2)。由于大、小体型猪种间可能存在除体型以外的差异,这些区间不一定都和体型大小相关。通过比较Z(H)较小区间和Z(Fst)较大区间之间的重叠区间来判定这些区间是否和体型大小相关(图1b)。为选择出极端候选受选择区域,使用242个Z(Fst)≥5的区间与Z(H)负值端最小的200个区间的重叠区域,再经合并得到13个候选区间(长度为240~720 kb)。这些候选区间共覆盖约4.2 Mb基因组区域,包含28个基因(图1a)。
a.猪18条常染色体上Z(Fst)正值端的分布,虚线为Z(Fst)=5;b.猪18条常染色体Z(H)负值端的分布,虚线为取200个最小Z(H)的阈值a.The positive end of the Z(Fst) plotted on 18 pig autosomes.A dashed horizontal line indicates the cut-off Z(Fst)=5;b.The negative end of the Z(H) plotted on 18 pig autosomes.A dashed horizontal line indicates the cut-off for 200 smallest Z(H)图1 常染色体上Z(Fst)和Z(H)的分布情况Fig.1 The distribution of Z(Fst) and Z(H) on autosomes
2.2相关性状QTL 在候选受选择区域的富集
为验证所得到的候选受选择区域和猪的生长、体重、体长等性状相关,在pig QTLdb数据库中搜索13个候选受选择区域所包含的已知QTL(http://www.animalgenome.org)。共发现924个QTLs与候选区间重叠,其中407个(~44%)QTLs与体型大小直接或间接相关。与生长、体型直接相关的QTLs为95个,如与体重、胴体长、胴体重、体长、脊椎数、日增重等相关QTLs(表2)。同时,大多数选择区域至少对应3个以上和生长相关的QTLs。这些结果说明和体型相关的QTL在候选区域富集,同时说明本研究检测选择信号的方法可靠。
表213个选择区域
Table 213 regions of selection

染色体Chromosome受选择区间RegionZ(Fst)Z(H)相关数量性状位点MappedQTL*1151180001~1514200007.040-3.940Bodyweight(20weeks)QTL(5)TeatnumberQTL(3)1153360001~1536200009.624-4.046Bodyweight(20weeks)QTL(5)DailyfeedintakeQTL(1)1256760001~2570600009.745-4.067AveragedailygainQTL(10)BodyweightQTL(4)1274500001~2747400007.415-3.940averagedailygainQTL(12)BodyweightQTL(7)278540001~787800005.298-3.452CarcassweightQTL(4)BodyweightQTL(2)846180001~469000008.442-3.919TeatnumberQTL(2)Bodyweight(birth)QTL(5)854900001~5532000010.247-4.088BodyweightQTL(5)FeetweightQTL(1)856080001~5642000011.768-4.322TeatnumberQTL(3)BodyweightQTL(5)865840001~660800009.667-4.025Bodyweight(birth)QTL(5)CarcasslengthQTL(1)891160001~914800005.325-3.322BodyweightQTL(3)FeetweightQTL(1)8102460001~1027000008.973-3.982Bodyweight(30weeks)QTL(2)HamweightQTL(1)13114200001~1144800007.284-4.004AveragedailygainQTL(4)VertebranumberQTL(1)1586720001~8706000010.779-4.088AveragedailygainQTL(2)Bodyweight(weaning)QTL(1)
* 括号中数字为相应QTL出现的次数
* The number in bracket indicate occurrence of the corresponding QTL
2.3候选基因的GO富集分析和通路分析
利用在线UCSC genome browser工具对候选区域进行基因注释,共发现了28个基因(图1a)。为阐明这28个基因的功能,使用PANTHER (Protein ANalysis THrough Evolutionary Relationships)对其进行功能注释及GO分析。结果表明,17个基因与机体生长发育直接或间接相关(表3)。这17个基因大多参与细胞生长发育的生物学过程或者重要基础信号通路。比如,IGF1R、MCTS1、OBSCN、NPY5R、SMC2等基因参与细胞生长、增殖、迁移、分化和凋亡[11];DNAJB6、NAF1、ZNF423等基因调节基础转录、翻译;IGF1R、CXXC4、ZNF423、TRIML1等基因参与调控动物体型大小的重要基础信号通路。
与动物体型大小相关的信号通路有:胰岛素信号通路、Wnt信号通路和Notch信号通路。胰岛素信号通路(亦称生长信号通路),调控个体发育始于胰岛素与胰岛素受体的结合,并由此引发细胞内一系列信号转导,最终到达各效应器官发挥作用。IGF1R不仅与细胞生长增殖直接相关,同时在胰岛素通路中起着关键调节作用,当IGF1R受体因子失活时,会抑制胰岛素信号通路功能的发挥,从而影响细胞、器官乃至个体大小。Wnt信号通路在生物进化过程中高度保守,该通路的开启或关闭控制着大量生长和代谢相关基因的表达[12]。而Notch信号通路是通过相邻细胞之间的相互作用来调节细胞、组织、器官的分化和发育,相邻细胞借助Notch受体与配体结合传递Notch信号,从而扩大并固化细胞间的分子差异,最终决定细胞命运。Notch信号通路影响器官形成和细胞正常形态,同时在调节细胞生长和维持组织体内平衡的过程中也起着十分关键的作用[13]。采用DAVID 软件对这些候选基因进行KEGG通路分析,结果只富集到了“Long-term depression”信号通路。从文献上看,该通路和神经突触的功能相关,并不与生长发育有直接联系。
表3生长发育相关基因的GO富集分析
Table 3GO term enrichment for genes related to growth and development

染色体Chromosome基因Gene生物学过程Biologicalprocess1MCTS1regulationoftranscription(GO:0006355);cellcycle(GO:0007049);positiveregulationofcellproliferation(GO:0008284);regulationofgrowth(GO:0040008);translation(GO:0006412);translationalinitiation(GO:0006413)1PGPEP1Lproteolysis(GO:0006508)1IGF1Rpositiveregulationofcellproliferation(GO:0008284);insulinreceptorsignalingpathway(GO:0008286);insulinreceptorsignalingpathway(GO:0008286);negativeregulationofap-optoticprocess(GO:0043066);organmorphogenesis(GO:0009887)1SMC2mitoticcellcycle(GO:0000278);cellcycle(GO:0007049)1VPS13AproteinretentioninGolgiapparatus(GO:0045053)2OBSCNpositiveregulationofGTPaseactivity(GO:0043547);multicellularorganismaldevelopment(GO:0007275);positiveregulationofapoptoticprocess(GO:0043065);apoptoticsignalingpathway(GO:0097190);celldifferentiation(GO:0030154)2DNAJB6negativeregulationoftranscription,DNA-templated(GO:0045892)4TRIML1multicellularorganismaldevelopment(GO:0007275);negativeregulationofWntsignalingpathway(GO:0030178)8GUCY1A3cGMPbiosyntheticprocess(GO:0006182);translationalinitiation(GO:0006413)8NPY1RG-proteincoupledreceptorsignalingpathway(GO:0007186);celldifferentiation(GO:0030154)8NAF1rRNAprocessing(GO:0006364);ribosomebiogenesis(GO:0042254)8CXXC4zygoticspecificationofdorsal/ventralaxis(GO:0007352);negativeregulationofapoptoticprocess(GO:0043066);negativeregulationofWntsignalingpathway(GO:0030178)8ZNF423Notchsignalingpathway(GO:0007219);celldifferentiation(GO:0030154);positiveregula-tionofBMPsignalingpathway(GO:0030513);regulationoftranscription,DNA-templated(GO:0006355);multicellularorganismaldevelopment(GO:0007275)8GUCY1B3cGMPbiosyntheticprocess(GO:0006182)8NPY5Rnegativeregulationofapoptoticprocess(GO:0043066);positiveregulationofsmoothmusclecellproliferation(GO:0048661)15METTL8methylation(GO:0032259)15DCAF17nucleolus(GO:0005730);integralcomponentofmembrane(GO:0016021);Cul4-RINGE3ubiquitinligasecomplex(GO:0080008)
综上推测,控制小型猪发育的基因很可能不局限在一个信号通路里,也可能不是由少量几个突变造成的,而是由多个参与细胞生长发育及生命基础代谢的基因突变来协同完成。
2.4候选基因的突变注释
对所有候选区间SNP和短插入缺失突变进行注释,共发现139个位于基因编码区及剪切位点的突变。其中,55个突变可能影响相关候选基因的蛋白质结构,包括50个错义突变,3个移码突变,2个剪切位点突变,其余全部为同义突变。3个移码突变都只出现在一个个体上,不太可能是影响小型猪的突变。两个剪切突变分别在TDO2和HIGH基因,两个突变在大小型猪群中都存在,且频率类似。在50个错义突变中,有5个突变在大、小型猪中有显著的频率差异,即大型猪以参考基因组(杜洛克猪)等位基因纯合子为主,小型猪以突变型纯合子为主(表4)。它们分别位于ENSSSCG00000027431、VPS13A、OBSCN、ENSSSCG00000027713和DCAF17基因。VPS13A调节蛋白等大分子的合成和运输,能够促进蛋白前体的吸收和转化,从而促进或抑制细胞增殖及其分化[14];OBSCN(Obscurins)编码骨架蛋白,在横纹肌中起着结构和调节作用;DCAF17介入CUL4-DDB1泛素酶调控通路,泛素酶通过对一些特定调节子的泛素化来调控细胞的增殖、存活、DNA修复和基因组完整性[15]。而ENSSSCG00000027431和ENSSSCG00000027713两个基因的功能未知。但这5个错义突变都处于基因的非保守区,表明其不太可能为导致大、小体型猪差异的突变位点。而IGF1R基因对发育过程中的细胞增殖、细胞迁移和器官形成等方面都起到重要的正向调控作用,同时参与调控动物体型大小的胰岛素信号通路,可作为本研究理想的候选基因,本研究在该基因上仅找到一个错义突变,但该突变等位基因仅在大体型猪群中以低频存在。鉴于本研究的限制,我们不排除有大的结构突变(CNV、大片段插入缺失等)或者调控序列突变,从而影响这些基因及候选区间内其他基因的表达,进而影响体型发育。
表4候选错义突变位点
Table 4Candidate missense mutations
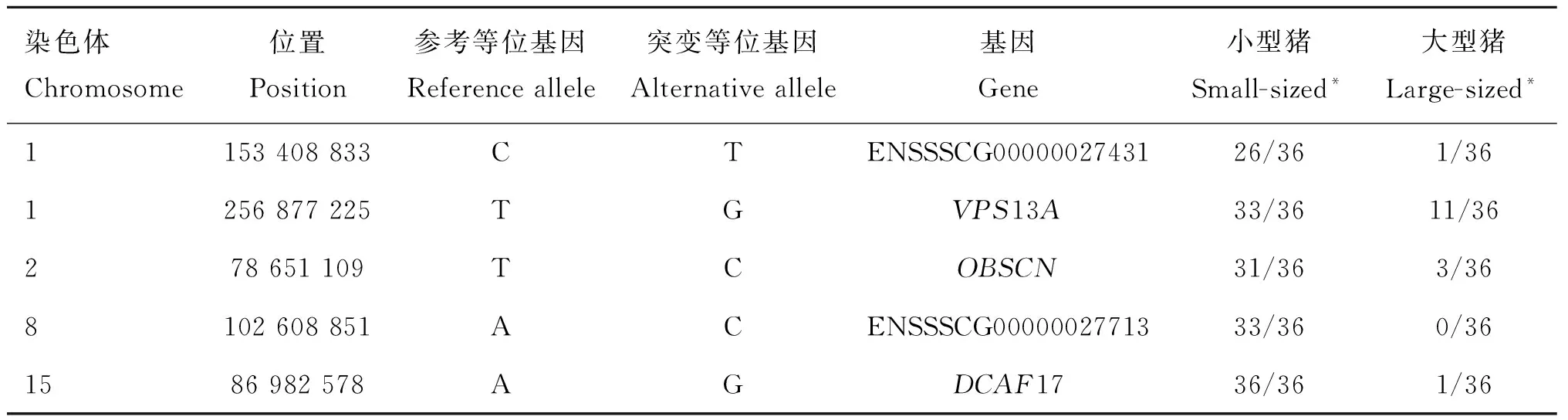
染色体Chromosome位置Position参考等位基因Referenceallele突变等位基因Alternativeallele基因Gene小型猪Small-sized*大型猪Large-sized*1153408833CTENSSSCG0000002743126/361/361256877225TGVPS13A33/3611/36278651109TCOBSCN31/363/368102608851ACENSSSCG0000002771333/360/361586982578AGDCAF1736/361/36
*. 突变等位基因在大、小体型猪群中的频率
*. The alternative allele frequency in small-sized and large-sized pigs
3 讨 论
近年来,第二代高通量测序技术在基因组学和遗传学中广泛应用,使得利用少量个体基因组测序信息检测基因组范围内的选择信号(Signature of selection)成为可能。大量与驯化、适应性和重要性状相关的受选择区段被报道,其中,涉及的研究对象有鸡、牛、狗、猪、羊等。本研究借鉴前人的策略,将五指山猪和巴马香猪合为小型猪组,将金华猪、二花脸猪和河套大耳猪合为大型猪组,在大、小体型猪分组间应用群体分化系数(Fst)初步确定两个分组间受选择区域。考虑到单个SNP的选择信号在经历正向选择时,通常会受到种群历史的随机因素影响,很难精确区分真实信号和噪音信号,故本研究结合40 kb 滑动窗口(Sliding window)遗传分化系数Fst和杂合度来确定体型相关的候选区域。
本研究共确定13个选择区域,包含了大量影响猪体重、胴体重、腿重和脊椎数的QTLs,同时,还包含了一系列影响猪采食量和平均日增重的QTLs[16]。此结果表明,小型猪的形成可能是从遗传上控制其身体及各个脏器的生长发育,同时导致其采食量的降低。此外,这13个区间共包含28个基因,通过PANTHER进行基因注释和富集分析,发现其中17个基因与机体的生长发育存在直接或者间接的关联。其中,IGF1R在哺乳动物的生长发育过程中起着重要作用。N.B.Sutter等鉴别出决定狗体型大小的关键基因IGF1[17],而IGF1主要是通过结合胰岛素样生长因子受体IGF1R和IGF2R,介导生长激素的促生长作用,通过自身磷酸化激活PI3K以及MAPK信号通路,调控细胞的增殖、分化和凋亡。此外,有研究表明,IGF1R缺陷小鼠在一定程度上患有侏儒症,同时会改变软骨细胞增殖,导致骨延长区生长板过度肥大并凋亡[18],在老鼠中将IGF1R敲除,结果表明IGF1会影响骨细胞介导的骨矿化,而骨矿化则是骨骼形成的关键,因此IGF1R可能是影响猪体型大小的关键候选基因。此外,候选基因GUCY1A3是蛋白编码基因,编码可溶性鸟苷酸环化酶(sGC)α亚基,而sGC是一氧化氮(NO)的主要受体,它启动NO信号转导通路,是NO-cGMP通路中的关键酶,它参与血管舒缩、抑制血小板凝集及细胞增殖和凋亡,是控制冠心病和动脉粥样硬化的重要因子,它在金属离子辅助因子如Mg2+或Mn2+存在下,可转换5在-三磷酸甘氨酸(三磷酸鸟苷)为3′,5′-磷酸鸟苷(cGMP)和焦磷酸。第二信使cGMP通过刺激蛋白激酶G可改变cGMP-gated离子通道和cGMP调节磷酸二酯酶活性,它的关键生理作用是调节细胞增生、细胞肥大、迁移、细胞外基质生产和细胞分化等过程[19]。研究表明,GUCY1A3的敲除会大幅度影响cGMP调节磷酸二酯酶活性,从而影响体内细胞的增殖分化。虽然目前有关GUCY1A3与动物体型大小的研究还不多,但GUCY1A3具有调控细胞增殖和凋亡的功能,使其可以作为影响体型大小较为理想的候选基因。另外,DCAF17介入CUL4-DDB1泛素酶调控通路, 泛素酶通过对一些特定调节子的泛素化来调控细胞的增殖、存活、DNA修复和基因组完整性。DCAFs(DDB1和CUL4相关因子家族)作为DDB1和CUL4蛋白直接的桥梁而介入细胞泛素化过程,从而调控细胞的基础功能[15],具有调控生长的可能。
本研究在受选择区域里富集的基因大多在生命体发育、基础代谢中起着极为关键的作用,多个基因还参与到Wnt、Notch等重要信号通路。这些关键基因上如果出现较大的蛋白质结构突变,有可能会导致个体死亡,这也从侧面解释了为什么候选基因中出现的突变大多在非保守区。同时也表明,五指山猪和巴马香猪小体型的形成不太可能是某个发育过程中关键基因的结构突变造成,而更可能是由于一个或者多个基因调节突变(比如位于启动子、增强子区域等)协同产生。类似的,M.Carneiro等研究发现,野兔的驯化过程中,野兔和家兔之间的行为差异主要来源于基因调控区域突变(研究人员并未发现驯化过程中有基因失活或主效基因的突变,驯化带来的改变主要发生在基因组的非编码区域)[20]。基因调控区的注释目前在人和小鼠的研究中依据其相应ENCODE项目产生的数据来实现[21]。当前,国际动物遗传学界开展的FAANG(Functional Annotation of Animal Genomes)计划将有类似ENCODE数据产生[22],该计划将在多个动物物种中系统进行基因表达谱、染色质开放区、调控序列(启动子、增强子、绝缘子等)、甲基化位点和三维染色质折叠调控的研究。届时,将有可能通过这些数据对猪的调控突变进行注释。此外,在本研究中,不能排除存在拷贝数变异或其他染色体结构突变在小型猪发育中起作用。
4 结 论
本研究基于全基因组重测序数据,结合种群遗传分化指数Fst和小型猪种内杂合度,进行系统选择信号检测,共确定了13个选择区域,包含28个基因。通过PANTHER进行基因注释和富集分析,发现其中17个基因与机体的生长发育存在直接或者间接的关联。GO基因富集分析鉴别到多个体型相关的基因,其中包含IGF1R、GUCY1A3等基因。受猪基因组注释不完善的影响,尤其欠缺调控序列的注释,本研究没有揭示造成小型猪形成的分子机制。后续通过完善基因及其调控序列的注释,将有助于解释形成小型猪体型的遗传变异。
致谢:感谢江西农业大学黄路生院士在试验设计和文章修改上提供指导。
[1]商海涛,魏泓.我国小型猪品系资源状况初浅分析[J].中国实验动物学报,2007,15(1):70-75.
SHANG H T,WEI H.Preliminary analysis of Chinese miniature pig strains and resources[J].ActaLaboratoriumAnimalisScientiaSinica,2007,15(1):70-75.(in Chinese)
[2]王峰,魏立民,郑心力,等.海南五指山猪生长发育性能测定试验[J].猪业科学,2009,26(5):108-109.
WANG F,WEI L M,ZHENG X L,et al.The analysis of growth development in Wuzhishan pig in Hainan[J].SwineIndustryScience,2009,26(5):108-109.(in Chinese)
[3]朱荣生,王怀中,张印.杜洛克和大约克猪体重和体尺性状间典型相关分析[J].家畜生态学报,2009,30(2):26-28.
ZHU R S,WANG H Z,ZHANG Y.Canonical correlation analysis of body weight and body measurement traits in duroc and yorkshire[J].ActaEcologiaeAnimalisDomastici,2009,30(2):26-28.(in Chinese)
[4]RUBIN C J,ZODY M C,ERIKSSON J,et al.Whole-genome resequencing reveals loci under selection during chicken domestication[J].Nature,2010,464(7288):587-591.
[5]AXELSSON E,RATNAKUMAR A,ARENDT M L,et al.The genomic signature of dog domestication reveals adaptation to a starch-rich diet[J].Nature,2013,495(7441):360-364.
[6]MOON S,KIM T H,LEE K T,et al.A genome-wide scan for signatures of directional selection in domesticated pigs[J].BMCGenomics,2015,16:130.
[7]刘真,王慧华,刘瑞凿,等.不同尾型绵羊全基因组选择信号检测[J].畜牧兽医学报,2015,46(10):1721-1732.
LIU Z,WANG H H,LIU R Z,et al.Genome-wide detection of selection signatures of distinct tail types in sheep populations[J].ActaVeterinariaetZootechnicaSinica,2015,46(10):1721-1732.(in Chinese)
[8]AI H S,FANG X,YANG B,et al.Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing[J].NatGenet,2015,47(3):217-225.
[9]MCKENNA A,HANNA M,BANKS E,et al.The genome analysis toolkit:a MapReduce framework for analyzing next-generation DNA sequencing data[J].GenomeRes,2010,20(9):1297-1303.
[10]COCKERHAM C C,WEIR B S.Covariances of relatives stemming from a population undergoing mixed self and random mating[J].Biometrics,1984,40(1):157-164.
[11]WANG Z,LU P,LIANG Z,et al.Increased insulin-like growth factor 1 receptor (IGF1R) expression in small cell lung cancer and the effect of inhibition of IGF1R expression by RNAi on growth of human small cell lung cancer NCI-H446 cell[J].GrowthFactors,2015,33(5-6):337-346.
[12]XU Y,YANG Z,YUAN H,et al.PCDH10 inhibits cell proliferation of multiple myeloma via the negative regulation of the Wnt/beta-catenin/BCL-9 signaling pathway[J].OncolRep,2015,34(2):747-754.
[13]KIDD S,LIEBER T.Mechanism of notch pathway activation and its role in the regulation of olfactory plasticity in drosophila melanogaster[J].PLoSOne,2016,11(3):e0151279.
[14]HONISH S,YU W,LIU G,et al.Chorein addiction in VPS13A overexpressing rhabdomyosarcoma cells[J].Oncotarget,2015,6(12):10309-10319.
[15]LEE J,ZHOU P.DCAFs,the missing link of the CUL4-DDB1 ubiquitin ligase[J].MolCell,2007,26(6):775-780.
[16]DOUMAYROU J,THEBAUD G,VUILLAUME F,et al.Mapping genetic determinants of viral traits with FST and quantitative trait locus (QTL) approaches[J].Virology,2015,484:346-353.
[17]SUTTER N B,BUSTAMANTE C D,CHASE K,et al.A single IGF1 allele is a major determinant of small size in dogs[J].Science,2007,316(5821):112-115.
[18]LEE Y,WANG Y,JAMES M,et al.Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells[J].MolCarcinog,2016,55(5):991-1001.
[19]SAINO M,MARUYAMA T,SEKIYA T.Inhibition of angiogenesis in human glioma cell lines by antisense RNA from the soluble guanylate cyclase genes GUCY1A3 and GUCY1B3[J].OncolRep,2004,12(1):47-52.
[20]CARNEIRO M,RUBIN C J,DI PALMA F,et al.Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication[J].Science,2014,345(6200):1074-1079.
[21]CONSORTIUM E P.An integrated encyclopedia of DNA elements in the human genome[J].Nature,2012,489(7414):57-74.
[22]ANDERSSON L,ARCHIBALD A L,BOTTEMA C D,et al.Coordinated international action to accelerate genome-to-phenome with FAANG,the functional annotation of animal genomes project[J].GenomeBiol,2015,16:57.
(编辑郭云雁)
Identifying Signatures of Selection Related to Small Body Size in Pigs
LI Wan-bo*#,ZHU Ya-ling#,AI Hua-shui,GUO Tian-fu*
(StateKeyLaboratoryforPigGeneticImprovementandProductionTechnology,JiangxiAgriculturalUniversity,Nanchang330045,China)
In this study,we analyzed genetic differentiation between small- and large-sized Chinese indigenous pigs,aiming to detect signatures of selection and to identify candidate genes and mutations related to small body size.Fixation index (Fst) and heterozygosity were calculated based on whole-genome resequencing data from miniature pigs (Wuzhishan and Bamaxiang) and large-sized pigs (Jinhua,Erhualian and Hetao).The regions with largerFstvalues and lower heterozygosity were regarded as candidate regions of selection.Through further annotation on gene function,pathway and mutations of the genes located in the candidate regions,we sought to screen genes and mutations related to body size.In total,32 475 218 SNPs were obtained after stringent filtering from whole-genome resequencing data.Fstand heterozygosity were calculated using 40 kb sliding window strategy.And 242 putative selection regions were kept by applying a Z-transformedFst(Z(Fst))>5.There are 13 regions overlapped between these 242 regions and the 200 regions with lowest Z-transformed heterozygosity (Z(H)),encompassing 28 genes.Seventeen out of the 28 genes are found to be associated with growth or body size through PANTHER annotation.Among these genes,IGF1RandGUCY1A3 were the most promising candidate genes.Together,by applying population genetic methods,we can detect regions of signature of selection related to body size in pigs.And after comprehensive analysis in gene function and mutations,we proposed that mutation(s) associated with small-sized phenotype in Wuzhishan and Bamaxiang pigs might be located in the non-coding regions detected.
miniature pigs;whole-genome resequencing;signatures of selection;body size;candidate genes
10.11843/j.issn.0366-6964.2016.10.005
2016-01-11
国家自然科学基金(31402058);江西省自然科学基金(2010GQN0045)
李完波(1982-),男,湖南岳阳人,助理研究员,博士生,主要从事生物信息学研究,Tel:0791-83813080,E-mail:li.wanbo.jxau@gmail.com;朱亚玲(1993-),女,江西上饶人,硕士生,主要从事动物遗传育种研究,Tel:0791-83813080,E-mail:yaling_zhu @qq.com。二者并列为第一作者
李完波,E-mail:li.wanbo.jxau@gmail.com;郭添福,E-mail:guotianfu2001@163.com
S828.2
A
0366-6964(2016)10-1977-09
