超高效液相色谱-质谱联用法测定藏药萨热十三味鹏鸟丸中乌头碱类含量
2016-11-07魏文芝海平骆桂法
魏文芝,海平,骆桂法
(青海省食品药品检验所,青海西宁810016)
超高效液相色谱-质谱联用法测定藏药萨热十三味
鹏鸟丸中乌头碱类含量
魏文芝,海平,骆桂法
(青海省食品药品检验所,青海西宁810016)
目的建立测定萨热十三味鹏鸟丸中乌头碱、次乌头碱及新乌头碱含量的超高效液相色谱-质谱联用法。方法采用显微特征鉴别法鉴别成药中的铁棒锤,色谱柱为AcQuity UPLC BEH C18柱,流动相A为甲醇,流动相B为0.1%甲酸溶液,梯度洗脱,采用电喷雾离子源(ESI)、正离子扫描(ES+)及多反应监测(MRM)测定萨热十三味鹏鸟丸中乌头碱、次乌头碱及新乌头碱的含量。结果萨热十三味鹏鸟丸成药粉末中可见铁棒锤特征性伞状或盔帽状淀粉粒;乌头碱进样量在13.15~2 630.00 ng范围内与峰面积线性关系良好(r=0.999 4),平均回收率为97.34%,RSD=1.79%(n=9);次乌头碱进样量在32.06~3 205.89 ng范围内与峰面积线性关系良好(r=0.999 9),平均回收率为96.09%,RSD=1.94%(n=9);新乌头碱进样量在10.23~2 046.00 ng范围内与峰面积线性关系良好(r=0.999 3),平均回收率为95.94%,RSD=1.75%(n=9)。结论该显微鉴别法简单易操作,显微特征明显,结果易辨识;该乌头碱类含量测定方法结果准确,重复性好,可用于同时测定萨热十三味鹏鸟丸中3种乌头碱类物质的含量。
萨热十三味鹏鸟丸;显微鉴别;超高效液相色谱-质谱联用法;乌头碱;次乌头碱;新乌头碱
萨热十三味鹏鸟丸是藏医习用药物,由麝香、木香、藏菖蒲、铁棒锤、诃子、甘草膏等13味中药组方,具有消炎止痛、通经活络、醒脑开窍的功效,主要用于治疗由中风及“白脉病”引起的口眼歪斜、麻木瘫痪、四肢关节不利,以及脉管炎、腱鞘炎、麻风等[1]。其临床应用广泛,已入选青海省2012年基本药物目录。铁棒锤含有毒性生物碱类成分乌头碱、次乌头碱及新乌头碱,三者毒性均较大,乌头碱成人致死量为2~5 mg,从染毒到死亡只需8 min至4 h[2],乌头碱、次乌头碱和新乌头碱小鼠静脉注射的半数致死量(LD50)分别为0.12~0.27 mg/kg,0.477 mg/kg,0.1~0.137 mg/kg[3]。为保证制剂的安全性,笔者对萨热十三味鹏鸟丸成药中的铁棒锤及乌头碱、次乌头碱、新乌头碱进行了定性、定量考察。
1 仪器与试药
1.1仪器
OLYMPUS BX-51型显微成像仪(日本Olympus公司);Waters AcQuity-TQD型超高效液相色谱三重四级杆质谱联用仪(美国Waters公司);Sartorius BS 224S型电子天平、Sartorius CP 225D型电子天平(德国Sartorius公司);HY-2型调速多用振荡器(国华电器有限公司),现报道如下。
1.2试药
乌头碱对照品(批号为110720-200410),次乌头碱对照品(批号为110798-200805,含量为99.5%),新乌头碱对照品(批号为110799-200404),均由中国食品药品检定研究院提供;甲醇(色谱纯,Dikma公司);甲酸(优级纯,J&K Sciencific Ltd);异丙醇、乙醚、三氯甲烷(分析纯,天津百世化工有限公司)。试验所用药材由青海金诃藏药药业有限公司提供,经青海省食品药品检验所骆桂法主任药师鉴定,均为正品。萨热十三味鹏鸟丸样品信息见表1。

表1 萨热十三味鹏鸟丸样品信息
2 方法与结果
2.1铁棒锤显微鉴别
分别取铁棒锤药材粉末和萨热十三味鹏鸟丸样品粉末少许,置载玻片上,加水合氯醛封片,置显微镜下观察。结果,铁棒锤药材粉末与萨热十三味鹏鸟丸样品粉末中均可见铁棒锤特征性淀粉粒[4-5],呈盔帽状、伞状。见图1。
2.2乌头碱、次乌头碱、新乌头碱含量测定
2.2.1仪器工作条件
色谱条件:色谱柱为AcQuity UPLC BEH C18柱,采用流动相A为甲醇,B为0.1%甲酸溶液,梯度洗脱,程序见表2;流速:0.2 mL/min;柱温:30℃。
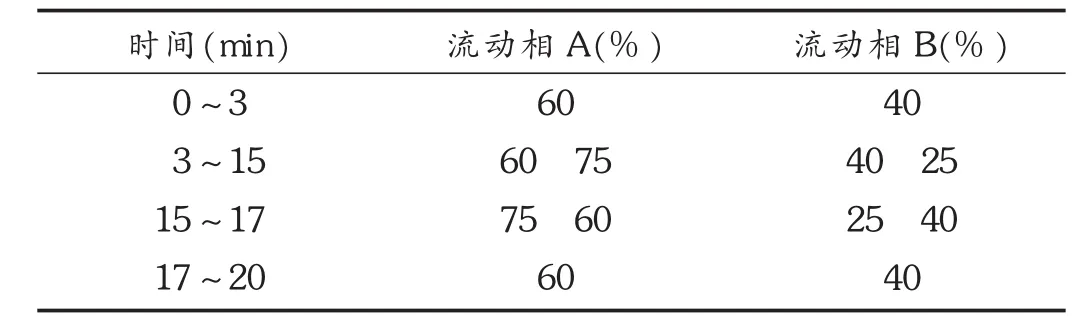
表2 梯度洗脱程序
质谱条件:电喷雾离子源(ESI),正离子扫描(ES+),多反应监测模式(MRM,见表3);毛细管电压4.0 kV;离子源温度140℃;去溶剂气温度250℃;去溶剂气流速480 L/h;锥孔气流速50 L/h;0~1 min,不进入质谱,1~3 min,进入质谱分析,3~20 min,不进入质谱。
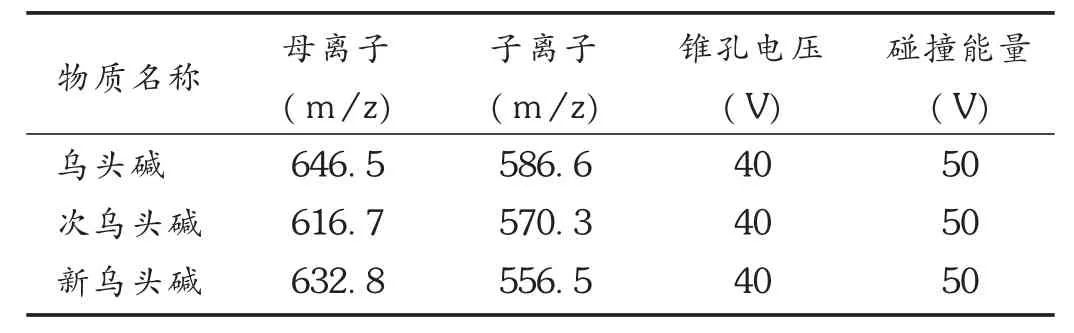
表3 MRM监测模式
2.2.2溶液制备
标准贮备液:分别精密称取乌头碱对照品13.15 mg、次乌头碱对照品16.20 mg及新乌头碱对照品20.72 mg,置100 mL容量瓶中,加异丙醇-三氯甲烷(1∶1)混合溶液适量,使溶解并稀释至刻度,摇匀,分别制成每1 mL中含乌头碱0.131 5 mg,次乌头碱0.161 1 mg,新乌头碱0.207 2 mg的标准贮备液。

图1 显微鉴别特征图
供试品溶液:取样品粉末约2.5 g,精密称定,置具塞锥形瓶中,加乙醚-三氯甲烷(1∶1)混合溶液50 mL,加氨试液6 mL,摇匀,放置过夜,滤过,滤渣加乙醚-三氯甲烷(1∶1)混合溶液50 mL,振摇1 h,滤过,滤渣用乙醚-三氯甲烷(3∶1)混合溶液洗涤3次,每次10 mL,合并滤液与洗液,70℃水浴蒸干,残渣加异丙醇-三氯甲烷(1∶1)混合溶液溶解,并定容至10 mL,经0.2 μm滤膜滤过,即得。
铁棒锤阴性对照溶液:按萨热十三味鹏鸟丸处方比例称取缺铁棒锤的阴性药材粉末2.5 g,精密称定,照供试品溶液制备方法制备,即得。
2.2.3方法学考察
专属性试验:精密吸取混合对照品溶液、供试品溶液及阴性对照溶液各5.0 μL,注入高效液相色谱仪,照2.2.1项下仪器工作条件试验。结果表明,其他成分对乌头碱、次乌头碱及新乌头碱测定无干扰,见图2。

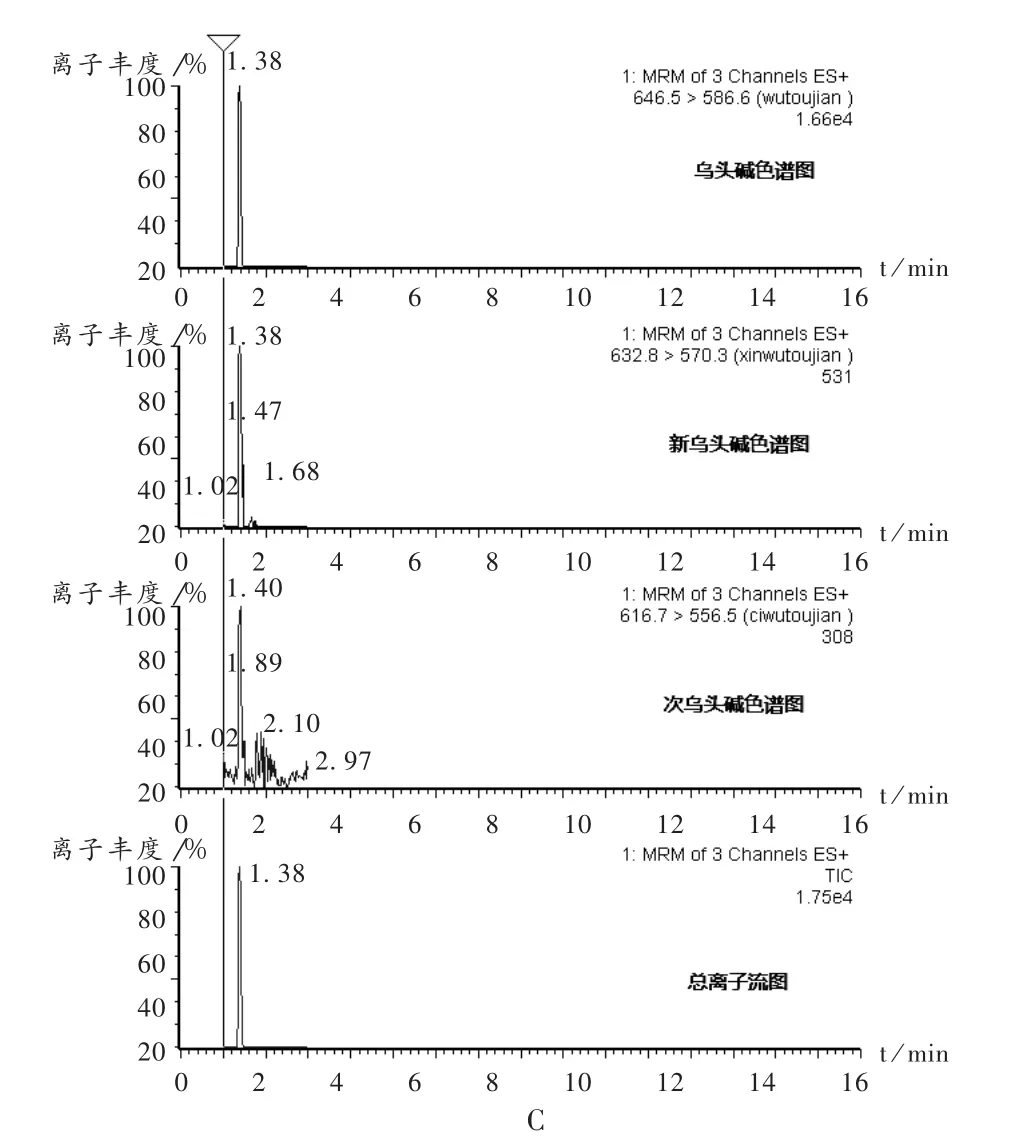
图2 高效液相色谱图
线性关系考察:精密量取乌头碱、次乌头碱及新乌头碱标准贮备液适量,制成混合对照品线性系列标准溶液(见表4)。分别吸取上述溶液5.0 μL,注入高效液相色谱仪,照2.2.1项下仪器工作条件试验,测定峰面积。以系列标准溶液中乌头碱、次乌头碱及新乌头碱的质量浓度(X,ng/mL)为横坐标、峰面积(Y)为纵坐标绘制标准曲线,得回归方程,乌头碱为Y=3.548 9 X+174.13,r=0.999 4(n=8),其进样量在13.15~263 0.00 ng范围内与峰面积线性关系良好;次乌头碱为Y=0.849 5X+ 19.629,r=0.999 9(n=8),其进样量在32.06~3 205.89 ng范围内与峰面积线性关系良好;新乌头碱为Y=0.323 9X+17.871,r=0.999 3(n=8),其进样量在10.23~2046.00ng范围内与峰面积线性关系良好。
精密度试验:精密吸取混合对照品溶液5.0 μL,照2.2.1项下仪器工作条件试验,连续进样6次。结果乌头碱、次乌头碱、新乌头碱峰面积的RSD分别为0.33%,0.89%,0.45%(n=6),表明仪器精密度良好。
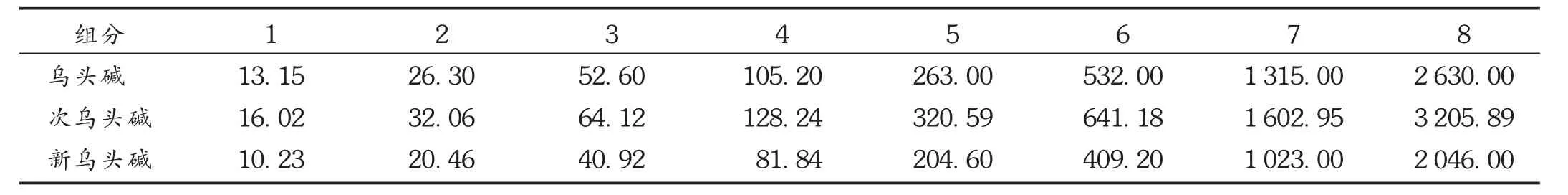
表4 混合对照品线性系列标准溶液质量浓度(ng/mL)
稳定性试验:精密吸取供试品溶液5.0 μL,分别于4,8,12,24 h时进样,测定峰面积。结果乌头碱、次乌头碱、新乌头碱峰面积的RSD分别为0.67%,0.84%,0.93%(n=4),表明供试品溶液在24 h内稳定。
重复性试验:取同一批样品(批号为100101),依法平行制备供试品溶液6份,照2.2.1项下仪器工作条件试验,测定乌头碱、次乌头碱及新乌头碱峰面积,计算乌头碱、次乌头碱及新乌头碱的含量。结果得乌头碱平均含量为8.996 4 μg/g,RSD为0.59%(n=6);次乌头碱平均含量为0.187 1 μg/g,RSD为1.06%(n=6);新乌头碱平均含量为1.691 7 μg/g,RSD为1.43%(n=6),表明方法重复性良好。
加样回收试验:精密称取同一批样品(批号为100101,已知乌头碱含量为8.996 4 μg/g)9份,各0.5 g,按相当于乌头碱含量的80%,100%,120%分别加入乌头碱对照品溶液2.7,3.0,3.5 mL,照2.2.1项下仪器工作条件试验,测定每份供试品中乌头碱的含量,计算回收率;精密称取同一批样品(批号为100101,已知次乌头碱含量为0.187 1 μg/g)9份,各1.5 g,按相当于次乌头碱含量的80%,100%,120%分别加入次乌头碱对照品溶液0.9,1.2,1.4 mL,照2.2.1项下仪器工作条件试验,测定每份供试品中次乌头碱的含量,计算回收率;精密称取同一批样品(批号为100101,已知新乌头碱含量为1.691 7 μg/g)9份,各0.5 g,按相当于新乌头碱含量的80%,100%,120%分别加入新乌头碱对照品溶液0.3,0.4,0.5 mL,照2.2.1项下仪器工作条件试验,测定每份供试品中新乌头碱的含量,计算回收率。结果见表5。
2.2.4样品含量测定
分别称取5批萨热十三味鹏鸟丸样品粉末2.5 g,照2.2.2项下方法制备供试品溶液,照2.2.1项下仪器工作条件试验,测定峰面积,计算乌头碱、次乌头碱及新乌头碱的含量。结果见表6。
3 讨论
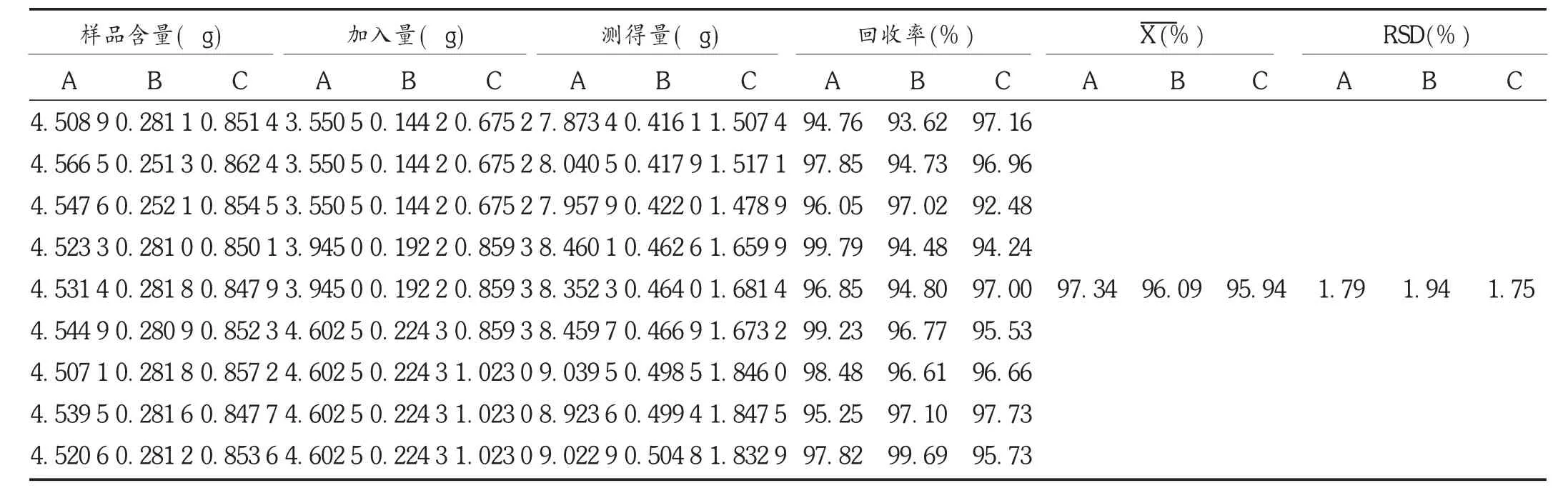
表5 加样回收试验结果(n=9)
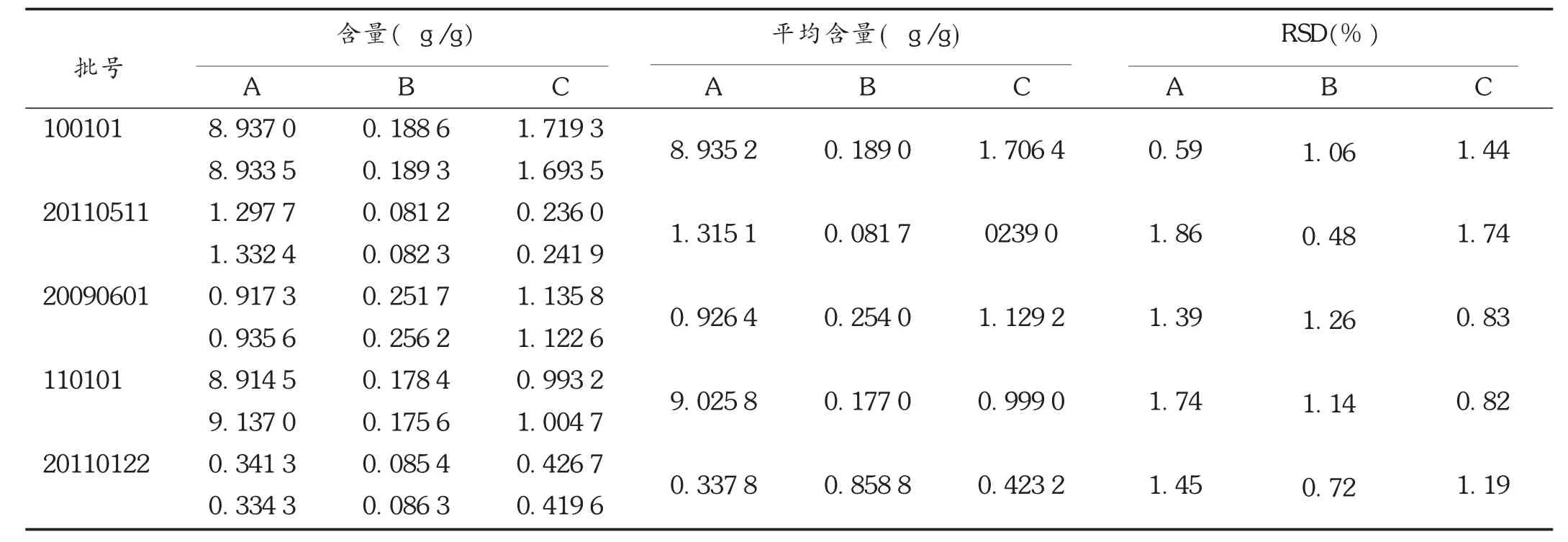
表6 样品含量测定结果(n=2)
[6-12],尝试多种乌头碱类物质的提取方法:1)供试品加0.1%氨溶液湿润后,加乙醚超声处理放置过夜后滤过,挥干,残渣加乙腈定容;2)供试品加氨试液湿润后,加乙醚振摇1 h,放置过夜后,滤过,挥干,残渣加三氯甲烷5 mL定容;3)供试品加氨试液,加异丙醇-乙酸乙酯(1∶1)混合溶液超声处理,滤液挥干,残渣加异丙醇-三氯甲烷(1∶1)混合溶液定容;4)供试品加乙醚-三氯甲烷(3∶1)混合溶液,加氨试液,放置过夜,滤过,滤渣70℃水浴蒸干,残渣加异丙醇-三氯甲烷(1∶1)混合溶液溶解并定容。试验结果证明,第4种方法提取效率最佳。
本试验结果显示,萨热十三味鹏鸟丸中乌头碱、次乌头碱、新乌头碱含量均较低,在安全范围内。本试验为其他含有铁棒锤药材的中藏成药中乌头碱类成分的含量考察提供了参考。
[1]WS3-BC-0330-95,卫生部药品标准·藏药(第一册)[S].
[2]陈冀胜,郑硕.中国有毒植物[M].北京:科学出版社,1987:465.
[3]周远鹏,江京莉.附子的研究——Ⅳ附子新乌头碱及其有关化合物的药理作用[J].中药药理与临床,1992,8(5):45.
[4]杨智锋,刘建锋,张红.铁棒锤药材质量标准研究[J].中国中药杂志,2005,30(22):1 771-1 773.
[5]胡本祥,李华,王宇鹏,等.铁棒锤的显微鉴别[J].西北药学杂志,2004,19(1):17-19.
[6]武洁,沈红,朱玲英,等.液-质联用法同时测定大鼠血浆中的乌头碱、新乌头碱、次乌头碱及其药动学[J].中国医院药学杂志.2011,31(14):1 162-1 166.
[7]邱葵,吴华,王鹤尧.LC-MS/MS方法同时测定人血浆和尿液中乌头碱与次乌头碱的含量[J].中国药学杂志,2010,45(8):633-636.
[8]Hayashida M,Hayakawa H,Wada K,et al.A column-switching LC/MS/ESI method for detecting tetrodotoxin and aconitum alkaloids in serum[J].Legal Medicine,2003,5(Suppl 1):101-104.
[9]Zhang F,Tang MH,Chen LJ,et al.Simultaneous quantitation of aconitine,mesaconitine,hypaconitine,benzoylaconine,benzoylmesaconine and benzoylhypaconine in human plasma by liquid chromatography-tandem mass spectrometry and pharmacokinetics evaluation of″SHEN-FU″injectable powder[J].J Chromatogr B Analyt Technol Biomed Life Sci,2008,873(2):173-179.
[10]包懿,宋凤瑞,刘志强,等.乌头碱类双酯型二萜生物碱水解反应的电喷雾质谱分析[J].质谱学报,2009,30(1):1-5.
[11]随志刚,姜雅秋,刘志强,等.乌头碱在家兔肠道内代谢产物的LC/ESI-MSn研究[J].化学学报,2009,67(21):2 439-2 444.
[12]索志荣,徐敏,秦海燕,等.LC-MS测定附子理中丸中3种双酯型生物碱含量[J].药物分析杂志,2010,30(12):2 279-2 282.
Content Determination of Aconitine,Hypaoconitine and Mesaconitine in SareShisanweiPengniao Pills by UPLC-MS
Wei Wenzhi,Hai Ping,Luo Guifa
(Qinghai Institute For Food and Drug Control,Xining,Qinghai,China810016)
ObjectiveTo establish an UPLC-MS method for the content determination of aconitine,hypaoconitine and mesaconitine in SareShisanweiPengniao Pills.MethodsThe microscopic identification method was used to identifythe aconitum in agents;the chromatographic column was AcQuity UPLC BEH C18column,mobile phase A was methanol and mobile phase B was 0.1%formic acid,gradient elution;the electrospray ionization(ESI),positive ion scan(ES+)and multiple reaction monitoring(MRM)were used todeterminethe contents of aconitine,hypaoconitine and mesaconitine in SareShisanweiPengniao Pills.ResultsThe linear relationship was good in the range of 13.15-2 630.00 ng for aconitine(r=0.999 4),and the average recovery rate was 97.34%,RSD=1.79%(n=9);the linear relationship was good in the range of 32.06-3 205.89 ng for hypaoconitine(r=0.999 9),and the average recovery rate was 96.09%,RSD=1.94%(n=9);the linear relationship was good in the range of 10.23-2 046.00 ng for mesaconitine(r=0.999 3),and the average recovery rate was 95.94%,RSD=1.75%(n=9).ConclusionThis microscopic identification method is simple and sensitive with significantly clear characteristics and easy to identify results;the UPLC-MS method is accurate and reliable with good repeatability,and can be used for the content determination aconitine,hypaoconitine and mesaconitine in SareShisanweiPengniao Pills.
SareShisanweiPengniao Pills;microscopic identification;UPLC-MS;aconitine;hypaoconitine;mesaconitine
R284.1;R286.0
A
1006-4931(2016)18-0025-05
青海省科技厅资助项目,项目编号:2012-N-523。
魏文芝(1984-),女,主管药师,硕士研究生,研究方向为药物分析,(电话)0971-8247794(电子信箱)304436784@qq.com。
(2015-06-17)
