普拉格雷合成综述
2016-10-22万风云
万风云
(浙江工业大学药学院,浙江杭州310014)
医药化工
普拉格雷合成综述
万风云
(浙江工业大学药学院,浙江杭州310014)
作为新一代血小板P2Y12受体阻断剂普拉格雷,与噻氯匹啶、氯吡格雷相比,能更快速、更有效地抑制血小板聚集,因此研究普拉格雷全合成具有非常重要的意义,综述了抗血栓药物普拉格雷的全合成方法,按照起始原料和方法的不同有下面几条路线,并分别对路线加以叙述总结。
普拉格雷;抗血栓;合成;综述
0 前言
普拉格雷(Prasugrel),化学名为2-乙酰氧基-5(α-环丙基羰基-2-氟苄基)4,5,6,7-四氢噻吩并[3、2-c]吡啶;是由日本第一制药三共公司和美国礼来公司联合开发的新一代血小板二磷酸腺苷受体阻断剂。于2009年2月和2009年7月分别获得欧盟委员会和FDA批准上市,商品名为Effient,适应症为心力衰竭、中风、不稳定心绞痛等心脑血管疾病。它是一种前体药物,在体内经过代谢后形成活性分子,与血小板P2Y12受体结合而发挥抗血小板聚集的活性[1]。
随着人们生活水平的提高和生活节奏的加快,同时也没有合理的体育锻炼,越来越多的人,特别是老年人出现血栓的概率非常高。据世界卫生组织统计,全世界每年因疾病死亡的人数大约有5100万人,其中有30%的人是死于心脑血管疾病,因此,对抗血栓药物的研究具有非常重要的意义。
目前,文献[2-4]中报道关于普拉格雷的合成方法比较多,我们按照起始原料的不同可分为以下几种方法合成普拉格雷。

Prasugrel
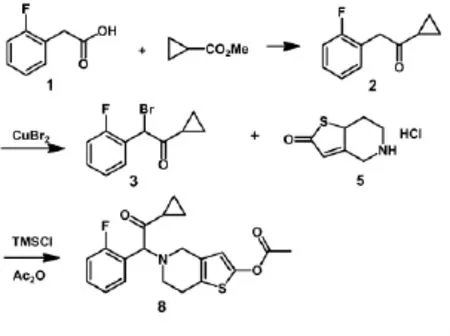
图1
1 以邻氟苯乙酸为原料
程兴栋等[5](如图1)以邻氟苯乙酸(1)和环丙基甲酸甲酯为原料合成1-环丙基-2-氟苄基酮(2),然后再经溴化铜溴代得到1-环丙基羰基-2-氟苄溴(3),化合物3再与5,6,7,7a-四氢噻吩并[3,2-c]吡啶-2(4H)-酮盐酸盐(5)缩合后乙酰化得到普拉格雷(8),总收率为72.9%。该工艺未使用高毒试剂或溶剂,操作简单,产率较高。但使用了1.5当量的溴化铜重金属,会严重污染环境,而且使用了格氏试剂异丙基溴化镁,操作繁琐且危险,工业化有一定的难度。
2 以1-环丙基-2-氟苄基酮为原料
竺伟等[6](如图2)用1-环丙基-2-氟苄基酮(2)在磺酰氯作用下顺利得到2-氯-1-环丙基-2-(2-氟苄基)乙酮(4),化合物4与5,6,7,7a-四氢噻吩并[3,2-c]吡啶-2(4H)-酮对甲苯磺酸盐(6)在碳酸钾、碘化钠作用下发生偶联反应得到7,化合物7在三乙胺作碱条件下经乙酰化反应得到普拉格雷(8),总收率为46%。这条路线使用了活性较低的氯代物(4),缩短了反应时间,减少了杂质的产生,使该步缩合反应收率提高,但路线总收率偏低。

图2
3 以邻氟溴苄为起始原料
孙志国等[8](如图3)以邻氟溴苄(9)和环丙基氰为原料,经格氏反应制得1-环丙基-2-氟苄基酮(2)再经NBS溴代得到1-环丙基羰基-2-氟苄溴(3),以N,N-二异丙基乙胺为缚酸剂,化合物3与5,6,7,7a-四氢噻吩并[3,2-c]吡啶-2(4H)-酮盐酸盐(5)通过缩合反应制得5-(α-环丙羰基-2-氟苄基)-2-氧-2,4,5,6,7,7a-六氢噻吩并[3,2-c]吡啶(7),化合物7经乙酰化得到普拉格雷(8),总收率为45%;该路线使用了剧毒物质环丙基氰,在反应过程中易产生氢氰酸等剧毒副产物,对人体的伤害极大,污染环境,且产率较低,是一条较老的工艺路线。
Pan X H等[7](如图4)也报道了以邻氟溴苄(9)作为起始原料,在四丁基溴化胺和氰化钠的作用下制得2-氟苯乙腈(10),化合物10经液溴溴代得到2-溴-(2-氟苯基)乙腈(11),化合物11与2-甲氧基-4,5,6,7a-四氢噻吩并[3,2-c]吡啶在碱性条件下反应得到2-(2-氟苯基)-2-(2-甲氧基-6,7二氢噻吩并[3,2-c]吡啶-5-(4H)-基)乙腈(13),13与环丙基溴化镁经格式反应得到1-环丙基-2-(2-氟苯基)-2-(2-甲氧基-6,7二氢噻吩并[3,2-c]吡啶-5-(4H)-基)乙酮(14),化合物14经盐酸水解和乙酰化作用得到普拉格雷(8),总收率为21%。这条路线使用了2-甲氧基-4,5,6,7a-四氢噻吩并[3,2-c]吡啶(12)作为缩合反应的一边侧链,用甲氧基保护了噻吩位的氧,提高了缩合反应收率和产物纯度。缺点是反应路线太长,而且使用了氰化钠,溴素等剧毒物,操作繁琐,收率十分低,成本较高,环境污染大,所以非常不适合工业化生产,但可作为学术研究推广。
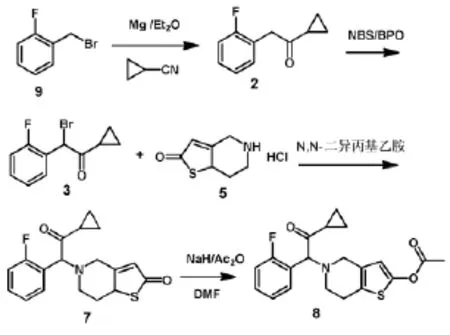
图3

图4
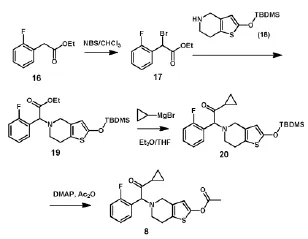
图5
4 以邻氟苯乙酸乙酯为起始原料
王相泉等[9](如图5)用邻氟苯乙酸乙酯(16)为起始原料,经N-溴代丁二酰亚胺溴化制得2-溴-2-邻氟苯基乙酸乙酯(17),化合物17与2-(叔丁基二甲基硅氧基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶(18)经缩合反应制得2-(叔丁基二甲基硅氧基)-5-(α-乙氧羰基-2-氟苄基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶(19),化合物19与溴化环丙基镁发生格氏反应得到2-(叔丁基二甲基硅氧基)-5-(α-环丙羰基-2-氟苄基)4,5,6,7-四氢噻吩[3,2-c]吡啶(20),化合物20经脱保护、乙酰化得到普拉格雷(8),总收率约为48%。此路线在制备化合物11时以羟基被保护的10为原料,可以避免O-位点的反应,减少杂质的产生,反应相对温和,但产率偏低,工业化应用成本较高。
5 总结
从本文中可看出普拉格雷的合成目前要经过格氏反应、溴代反应、缩合反应、脱保护乙酰化反应,工艺中难点不多,但反应步骤过长,操作繁琐,产率偏低,有待优化。在整个工艺中,缩合反应是关键一步,如1-环丙基羰基-2-氟苄溴与5,6,7,7a-四氢噻吩并[3,2-c]吡啶-2(4H)-酮盐酸盐间的缩合反应,文献中普遍使用碳酸钾和三乙胺作为缚酸剂,而且对1-环丙基-2-氟苄基酮上溴,然后脱溴缩合,不经反应步骤多,产率降低,有待优化;目前已有文献[10]报道通过催化量溴化铜一步法合成氯吡格雷,优化后降低了环境污染,缩短了反应步骤,提高收率,节约成本,值得借鉴来合成普拉格雷。
[1]荆亚萍,申东升,邓爵安,等.新一代血小板抑制剂-普拉格雷[J].国际药学研究杂志,2008,35(5):373-376.
[2]张雅然,郝晓燕,米国瑞,等.盐酸普拉格雷的合成工艺改进[J].中国新药杂志,2014,23(3):275-277.
[3]代黎,姜志辉,蔡瞻,等.盐酸普拉格雷合成工艺的改进[J].药学实践杂志,2013,31(3):195-197.
[4]李素义,梁艳霞,陈国华,等.盐酸普拉格雷合成路线图解[J].中国医药工业杂质2010,41(11):869-871.
[5]程兴栋,童玲,杨玉雷,等.普拉格雷的合成工艺研究[J].中国新药杂志,2010,19(15):1314-1316.
[6]竺伟,陈宇.普拉格雷的合成[J].中国医药工业杂志,2012,43(8):647-648.
[7]Pan X H,Huang R,Zhang JS,et al.Efficient synthesis of prasugrel,a novel P2Y12 receptor inhibitor[J].TetrahedronLetters,2012,53,5364-5366.
[8]孙志国,侯建,邹强,等.普拉格雷的合成[J].中国医药工业杂志,2009,40(4):244-246.
[9]王相泉,岳珊珊,李进都,等.普拉格雷的合成[J].中国医药工业杂志,2014,45(10):913-915.
[10]Ryan W E,Jason R Z,Zhu S L,et al.Simple catalytic mechanism for the direct coupling ofα-carbonyls with FuncTionalized Amines:A One Synthesis of Plavix[J]. 2013,135:16074-16077.
Review of the Synthesis of Prasugrel
WAN Feng-yun
(College of Pharmaceutical Science,Zhejiang University of Technology,Hangzhou,Zhejiang 310014,China)
As a new generation of platelet P2Y12 receptor antagonist prasugrel,compared with ticlopidine,clopidogrel,more quickly and more effectively inhibit platelet aggregation,therefore,the total synthesis of prasugrel has very important significance.This paper reports the antithrombotic drug prasugrel total synthesis,in accordance with the startingmaterials and differentmethods have the following route,and then to summarize the route.
Prasugrel;antithrombosis;synthesis;overview
1006-4184(2016)9-0004-03
2016-02-29
万风云(1988-),男,江西丰城人,从事药物及中间体的合成。E-mail:wanfengyun90@163.com。
