整合过表达嘌呤代谢途径关键酶基因提高酿酒酵母菌株环磷酸腺苷产量
2016-10-21王凯姬晓兵徐欢欢仇申珅邹少兰
王凯,姬晓兵,徐欢欢,仇申珅,邹少兰
(天津大学 化工学院,天津,300072)
整合过表达嘌呤代谢途径关键酶基因提高酿酒酵母菌株环磷酸腺苷产量
王凯,姬晓兵,徐欢欢,仇申珅,邹少兰*
(天津大学 化工学院,天津,300072)
调控嘌呤代谢途径关键酶基因表达水平,会影响经AMP→ADP→ATP 而生成的cAMP(cyclic adenosine mnophosphate,cAMP)产量。将酿酒酵母编码磷酸核糖焦磷酸合成酶基因PRS1和PRS3、磷酸核糖焦磷酸氨基转移酶基因ADE4、腺苷酸激酶基因ADK1,分别置于磷酸甘油酸激酶基因PGK1的启动子PGK1p和终止子PGK1t控制下,利用整合载体YIplac211,在菌株GA125中进行整合表达,通过摇瓶实验考察所得菌株的环磷酸腺苷产量。结果表明,在加腺嘌呤条件下,120 h时的cAMP产量分别为(5 380.5±1.91)、(5 189.4±1.72)、(5 131.8±2.05)和(5 199.9±1.62)μmol/L,较菌株GA125分别提高9.8%、5.9%、4.8%和6.2%;此外,ADK1过表达导致一定的生长抑制,所有菌株的腺嘌呤消耗量在(0.399±0.01)~(0.447±0.02)g/L,葡萄糖利用和乙醇产生上彼此没有呈现明显差异。
酿酒酵母;嘌呤代谢途径;整合;环磷酸腺苷;腺嘌呤
环磷酸腺苷(cyclic adenosine monophosphate, cAMP)是细胞内参与调节物质代谢和生物学功能的重要物质,它广泛参与机体内的各种生理活动,是生物体内重要的第二信使。 cAMP 不仅对于心脑血管疾病、恶性肿瘤、失眠健忘、再障性贫血等多种疾病均能达到好的治疗效果[1],在畜牧养殖业上也有重要的应用价值[2]。
在 cAMP 生产的主要3种方式——化学合成法、酶合成法和微生物发酵法中,发酵法具有可实现持续生产、工艺简单、能利用廉价的碳源、毒副作用小等多种优势[3]。而酿酒酵母(Saccharomycescerevisiae)不仅具有工业化应用技术成熟、抗逆性强、为食品安全性GRAS 微生物的优点,同时作为最早实现全基因组测序的微生物菌种,又是分子生物学和遗传学研究的最重要模式生物之一[4]。因此,通过遗传工程手段的改造实现以酿酒酵母为菌种的cAMP 发酵法生产,具有其特殊的优势。
酿酒酵母胞内嘌呤核苷酸的合成有从头合成途径(De Novo Synthesis)和补救途径(Salvage Pathway)(图1)[3]。研究证明,当胞内ADP、ATP浓度过高时,ADP、ATP通过反馈抑制从头合成途径第1个酶——磷酸核糖焦磷酸氨基转移酶(5-phosphoribosyl 1-pyrophosphate amidotransferase,PRPP amidotransferase)即Ade4p(对应基因为ADE4)的活性,而使从头合成途径基因的转录水平下降,表达受到抑制。当培养基中有相当量的腺嘌呤存在时,腺嘌呤进入胞内,通过补救途径形成AMP,AMP进一步转化为ADP和ATP,进而导致整个从头合成途径合成受到抑制[5-6]。催化AMP、ADP和ATP相互转化的腺苷酸激酶(adenylate kinase)由ADK1基因编码,是一种进化上高度保守的磷酸转移酶,可维持ATP、ADP和AMP间的平衡,在调节能量代谢和维持细胞内核苷酸的稳定方面有重要作用[7-8]。另一方面,作为机体中重要的代谢中间产物,PRPP参与包括嘌呤核苷酸从头合成途径和补救途径在内的细胞内多条代谢途径,其浓度受到严格控制;就嘌呤代谢途径而言,控制PRPP浓度的一个途径是通过终产物AMP、GMP反馈抑制磷酸核糖焦磷酸合成酶(PRPP synthetase)的活性。PRPP合成酶复合物亚基1~5分别由基因PRS1-PRS5编码,5个基因都能表达,但单个基因的表达产物都没有PRPP合成酶活性[9]。研究证明[10-11],缺失PRS2或PRS4基因对细胞生长代谢影响较小,而缺失PRS1或PRS3基因对细胞代谢影响显著;在缺失PRS1或PRS3的情况下,PRS5为细胞活性所必需。
由上述可知,PRPP合成酶、PRPP氨基转移酶Ade4p和腺苷酸激酶Adk1p都会影响到cAMP前体物ADP、ATP的合成,提高此3个关键酶基因的表达水平,预测可以提高cAMP的产量。姬晓兵等使用磷酸甘油酸激酶基因PGK的启动子PGK1p及终止子PGK1t,通过多拷贝游离型质粒YEplac195分别过表达PRS1、PRS3、ADE4和ADK1基因,结果PRS1、PRS3、ADE4过表达菌株的cAMP产量分别提高了9.03%、4.17%和6.06%,而ADK1过表达菌株则降低了3.85%[3]。为了提高基因表达的稳定性,同时观察拷贝数的影响,判断ADK1过表达菌株产量下降是否与ADK1过高的拷贝数和表达水平有关本研究改为通过整合载体YIplac211[12]分别单拷贝整合4个基因到染色体上,然后进行生长及cAMP发酵评价。
1 材料与方法
1.1材料
1.1.1菌株、质粒和引物
本实验所用质粒、菌株和引物见表1。
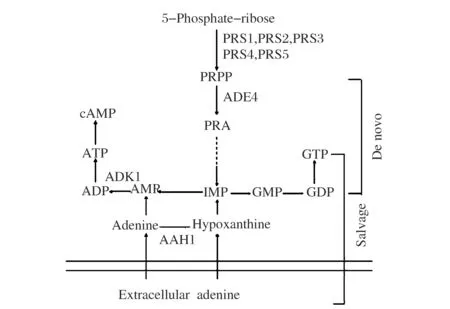
图1 酿酒酵母嘌呤核苷酸合成的部分途径Fig.1 Part of purine nucleotide synthesis pathway in S. cerevisiae

表1 实验所用质粒、菌株和引物
1.1.2培养基和培养条件
Luria-Bertani(LB)培养基(g/L):酵母抽提物 5,胰化蛋白胨 10,NaCl 10,pH 7.0,固体培养基则还需添加琼脂粉 15,用于转化子筛选时添加氨苄青霉素至终浓度100 mg/L,用于大肠杆菌的培养。固体培养条件:37 ℃ 培养箱倒置;液体培养条件:37 ℃、220 r/min。
YPD培养基(g/L):酵母抽提物10,蛋白胨20,葡萄糖20,自然pH值,固体培养基则还需添加琼脂粉15,用于酿酒酵母的培养。固体培养条件:30 ℃培养箱倒置;液体培养条件:30 ℃、220 r/min。
CMG-URA培养基(g/L):YNB 6.7,葡萄糖20,氨基酸碱基混合物0.83,固体培养基还需添加琼脂粉15,并调节pH至6.5;液体培养基调节pH至5.6。平板用于酿酒酵母转化子筛选,30 ℃静止培养。氨基酸碱基混合物的组成如下(mg/L):腺嘌呤50,亮氨酸100,精氨酸20,赖氨酸30,天冬氨酸100,甲硫氨酸20,谷氨酸100,苯丙氨酸50,组氨酸100,丝氨酸150,异亮氨酸30,苏氨酸150,色氨酸l00,酪氨酸30,缬氨酸150。
发酵培养基(g/L):酵母抽提物20,蛋白胨40,葡萄糖125,腺嘌呤1.25,自然pH值.用于酿酒酵母整合菌株的生长与发酵评价,30 ℃、220 r/min培养。
1.1.3主要试剂和设备
限制性内切酶购自美国Fermentas公司,PCR用酶购自北京全式金生物有限公司,T4连接酶购自美国Thermo Scientific公司;PCR产物和DNA片段切胶回收试剂盒购自北京博迈德生物技术有限公司;引物合成与基因测序由北京奥科鼎盛科技有限公司完成.氨苄抗生素与生化试剂购自上海生工生物工程股份有限公司,所用化学试剂为分析纯。
紫外可见分光光度计,美国UNICO公司;梯度PCR扩增仪,德国BIOMETRA公司;凝胶成像分析系统,美国Bio-rad公司Gel Doc XR;恒温培养箱,德国Memmert公司;Waters Alliance 2695高效液相色谱仪,美国Waters公司。
1.2实验方法
1.2.1分子生物学基本操作
大肠杆菌质粒DNA提取用碱裂解法[13]。酿酒酵母感受态细胞制备和转化用醋酸锂法[14]。其他有关操作如PCR、菌落PCR鉴定、酶切反应、琼脂糖凝胶电泳、连接、染色体提取等均参照文献[13,15]。
1.2.2四个整合质粒的构建
大量提取制备表1中的4个穿梭质粒,分别PstI和EcoRI双酶酶切后与同样双酶酶切的整合质粒YIplac211大片段连接,转化大肠杆菌DH5α感受态细胞,涂布添加了氨苄青霉素(终浓度100 mg/L)的LB平板,挑取平板生长菌落,接种添加了氨苄青霉素(终浓度100 mg/L)的LB液体培养基进行培养,提取质粒进行酶切鉴定,得到4个目的质粒YIplac211-PGK1p-PRS1-PGK1t、YIplac211-PGK1p-PRS3-PGK1t、YIplac211-PGK1p-ADE4-PGK1t和YIplac211-PGK1p-ADK1-PGK1t。
1.2.3整合菌株的构建
将上述4个整合质粒和对照质粒YIplac211分别首先用StuI单切线性化,然后用醋酸锂法[13]转化菌株GA125感受态细胞,用CMG-URA平板进行筛选,挑取平板生长菌落接种YPD液体培养基进行培养,提取染色体进行PCR鉴定,使用表2中的引物对P1、P4。这里,利用整合载体上的URA3基因被StuI酶切分开的两个片段分别作为左、右同源臂整合到GA125菌株染色体的URA3位点。
1.2.4整合菌株的生长与发酵评价
种子液培养:挑取固体培养基平板上生长的菌落,接入装有 5 mL YPD 培养液的试管中,30 ℃ 下 220 r/min 过夜培养,然后转接进行二次扩大培养,所得菌液为种子液。
发酵培养:新鲜种子液接种到装有25 mL发酵培养基的100 mL容量瓶中,控制初始OD600值在1左右,30 ℃、220 r/min下发酵,间隔12 h或24 h取样进行分析。
发酵液的分析:(1)OD600测定:将发酵样品适当稀释后测定 OD600值,检测生长情况;(2)HPLC 分析 cAMP 和腺嘌呤浓度:将发酵样品在 13 000 r/min、1 min 条件下离心,取上清进行适当稀释,用孔径 0.22 μm 的滤膜过滤,滤液用于色谱检测:检测波长 258 nm,Thermo Syncronis C18色谱柱,流动相组分为5.78 g/L KH2PO4,2.72 g/L 四丁基溴化铵(用磷酸调节 pH 至 4.3)与乙腈之体积比为85∶15,流速 1 mL/min,柱温 35 ℃。
所有发酵实验均进行了3次重复。
2 结果
2.1整合质粒的构建
本课题前期工作中,利用RT-qPCR方法比较了1.2.4所示发酵条件下菌株GA125细胞PGK1基因相对于PRS1、PRS3、ADE4和ADK1基因的转录水平,证明了PGK1基因启动子启动强度高于4个基因。故这里仍然选择使用PGK1基因的调控元件(PGK1p和PGK1t)来进行4个基因的表达。YIplac211和构建的4个整合质粒示意图见图2。

图2 YIplac211和YIplac211-PGK1p-ORF-PGK1t质粒示意图Fig.2 Schematic illustration of plasmids YIplac211 and YIplac211-PGK1p-ORF-PGK1t
4个整合质粒的PstI+EcoRI双酶酶切和StuI单酶酶切产物的琼脂糖凝胶电泳图谱见图3。
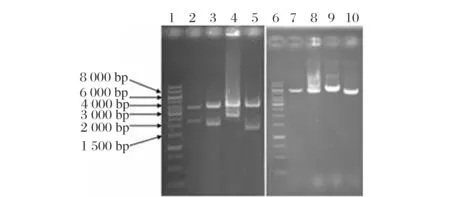
Lanes 1, 6:ladder; lanes 2-5:PstI + EcoRI, (3762+2252), (3762+ 1960), (3762+2530) and (3762+1666) bp for gene PRS1, PRS3, ADE4 and ADK1-containing integrating vectors, respectively; lanes 7-10:Stu I, 6014, 5722, 6292 and 5428 bp for gene PRS1, PRS3, ADE4 and ADK1-containing integrating vectors, respectively图3 整合质粒酶切产物琼脂糖凝胶电泳图谱Fig.3 Agarose gel electrophoresis of enzymatically digested products of integrating vectors
2.2整合菌株的构建
如图2所示,经StuI酶切线性化后,利用整合载体上的URA3基因被Stu I酶切分开的两个片段分别作为左、右同源臂整合到GA125菌株染色体的URA3位点.对CMG-URA平板筛选得到的菌株进一步进行PCR鉴定,以染色体DNA为模板、P1/P2为引物对的PCR鉴定结果见图4。

Lane 1:ladder; lanes 2-5:2252, 1960, 2530 and 1666 bp for gene PRS1, PRS3, ADE4 and ADK1-integrating strains, respectively图4 整合菌株PCR鉴定产物琼脂糖凝胶电泳图谱Fig.4 Agarose gel electrophoresis of PCR products for recombinant strains
2.3整合菌株的生长与发酵评价
对经过上述筛选、鉴定得到的整合菌株进行了摇瓶发酵实验,结果见图5。
由生长曲线可以看出:不同整合菌株的生长在24 h后彼此呈现出较为明显的差异,120 h时的OD600值分别为(81.8±0.16)(GA125-IP1)、(71.8±0.20)(GA125-IP3)、(80.0±0.14)(GA125-IE)、(62.3±0.12)(GA125-IK)和(70.0±0.11)(GA125-I),暗示了不同基因单拷贝整合过表达造成的生长效应不同。其中GA125-IK自48 h起OD600值低于对照菌株,表现出了一定程度的生长抑制。
由cAMP发酵曲线可以看出:4个基因的过表达均提高了cAMP的产量,120 h时的浓度分别为(5380.5±1.91)(GA125-IP1)μmol/L、(5189.4±1.72) (GA125-IP3)μmol/L、(5131.8±2.05) (GA125-IE)μmol/L、(5199.9±1.62)(GA125-IK)μmol/L和(4898.6±1.87) (GA125-I)μmol/L,增幅分别达9.8 %、5.9 %、4.8 %和6.2 %。
由腺嘌呤消耗(累计)量曲线可以看出:GA125-IP1和对照菌株GA125-I前期(72 h)消耗量低于其余三个整合菌株,72 h后GA125-I则稍稍高于所有整合菌株;菌株GA125-IP3、GA125-IE和GA125-IK之间则自始至终彼此出入不大。所有菌株的腺嘌呤最大消耗量在(0.399±0.01)~(0.447±0.02)g/L。
葡萄糖浓度曲线显示出所有菌株利用葡萄糖的速度差异不大,仅菌株GA125-IP3在后期利用速度上稍稍高于其他菌株;120 h时的残糖含量为(0.23±0.03)~(0.39±0.02)g/100 mL。而与葡萄糖利用能力相应,乙醇浓度曲线显示出所有菌株利用葡萄糖产乙醇的速度上彼此也出入不大,乙醇浓度在(48~72) h进入平台期,最大乙醇浓度在(2.69±0.04)~(3.00±0.02) g/100 mL。
综合分析上述各个指标之间的关系,发现其与4个基因类型间的关联并不明显。在4个基因单拷贝整合过表达都能提高cAMP产量的基础上,生长-cAMP产量-腺嘌呤利用能力-葡萄糖利用能力-产醇浓度之间的关联性,没有呈现出因基因种类变化而变化的明显的、一致的某种规律。

图5 整合菌株生长、cAMP发酵、腺嘌呤消耗、葡萄糖和乙醇浓度曲线Fig.5 The growth, cAMP fermentation, adenine consumption, glucose and ethanol curves of five integrating strains
3 讨论
酿酒酵母中cAMP的合成涉及相当复杂的代谢途径和调控机制。本研究结果初步证明了提高嘌呤代谢途径关键酶基因PRS1、PRS3、ADE4和ADK1的表达水平,确能提高cAMP产量。而每个基因提高cAMP产量的程度,及其过表达菌株的生长、腺嘌呤和葡萄糖利用能力、产乙醇特性等等,推测是下述因素综合作用的结果:(1) 基因功能在酿酒酵母整个代谢途径中的作用;(2) 出发菌株中基因的表达水平;(3) 在出发菌株中过表达基因的方式(由此决定了其过表达的水平和受到的调控机制);(4) 过表达菌株的培养和发酵条件。
本课题前期工作中,利用RT-qPCR方法比较了1.2.4所示发酵条件下菌株GA125细胞中若干基因的相对转录水平,发现PRS和ADE4中的基因最高转录水平也只有组成型表达基因PGK1转录水平的2.4%,而转录水平相对较高的ADK1,为PGK1转录水平的12.1 %。这个结果证明了PGK1启动子在本课题培养条件下仍然为强启动子,故本研究中使用PGK1的启动子和终止子来表达4个基因,预测能使这些基因以高于出发菌株野生型基因的水平组成型表达,cAMP产量的提高证明了这一点。
另一方面,对比本研究中单拷贝整合方式过表达和多拷贝游离型质粒形式过表达[3]的结果,反映出针对不同功能和不同基础表达水平的基因,合适的剂量至关重要。如上所述,ADK1基础表达水平相对远高于PRS1、PRS3和ADE4基因,故多拷贝游离型质粒载体可能导致其表达水平不适当地过高而产生某种不平衡,最终cAMP产量不升反降(较对照菌株下降3.85 %),而生长不受影响[3]。而PRS1、PRS3和ADE4基因单拷贝整合过表达的增产效应与多拷贝游离型质粒形式过表达的增产效应差异不大的结果则进一步暗示了PGK1强启动子下的单个拷贝数整合过表达可能已实现其增产潜力,或者存在其他的调控因素限制了高拷贝数表达下的增产效果。其中值得注意的一个问题是:多拷贝游离型质粒形式过表达菌株的生长、发酵评价体系没有提供维持质粒稳定存在的必需的选择压力,随着发酵的进行,可能存在的质粒丢失问题会影响到cAMP发酵动力学和最终产量。因此,要严格考察4个基因最大的增产潜力、进一步提高cAMP产生水平,在保证基因表达稳定性的前提下继续优化基因剂量即提高整合拷贝数的同时,还需要利用包括蛋白水平、代谢物水平在内的检测手段而加强对其它可能制约因素的分析和调控。
目前,酿酒酵母中生产cAMP的报道基本上都是基于cAMP 信号传导途径的基础研究[5-6,16],本实验室通过酿酒酵母发酵生产cAMP的报道尚属首次。本研究将有助于推动高值医药产品的绿色清洁生产,具有重要的应用意义。
[1]回皓升,梁高燕,吴铿.环磷腺苷治疗慢性充血性心力衰竭急性加重期的临床疗效研究[J].医师进修杂志, 2005,28(4):32-33.
[2]汪善锋,陈安国.环腺苷酸的生理功能及在动物生产中的应用[J].中国饲料,2003(10):3-5.
[3]姬晓兵,王凯,陈洵.过表达嘌呤合成途径关键酶基因对重组酿酒酵母菌株生产cAMP的影响[J].过程工程学报,2014,14(5):86-92.
[4]汪天虹.微生物分子育种原理与技术[M].北京:化学工业出版社,2005.
[5]REBORA K,DESMOUCELLES C,BOME F,et al.Yeast AMP pathway genes respond to adenine through regulated synthesis of a metabolic intermediate [J].Mol Cell Biol,2001,21(23):7 901-7 912.
[6]LJUNGDAHL P O,DAIGNAN-FOMIER B.Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae [J].Genetics,2012,190(3):885-929.
[7]ANNE-LISE V,JÖRG S.The adenylate kinase reaction acts as a frequency filter towards fluctuations of ATP utilization in the cell [J].Biophysical Chemistry,1987,26:19-28.
[8]DZEJA P P,TERZIC A.Phosphotransfer networks and cellular energetics [J].Journal of Experimental Biology,2003,206:2 039-2 047.
[9]HOVE-JENSEN B.Heterooligomeric phosphoribosyl diphosphate synthase ofSaccharomycescerevisiae:combinatorial expression of the fivePRSgenes inEscherichiacoli[J].J Biol Chem,2004,279(39):40 345-40 350.
[10] CARTER AT,BEICHI F,HOVE-JENSEN B,et al.PRS1 is a key member of the gene family encoding phosphoribosylpyrophosphate synthetase inSaccharomycescerevisiae[J].Mol Gen Genet,1997,254(2):148-156
[11]YOLANDA H,ADRIAN P,MICHAEL S.PRS5, the fifth member of the phosphoribosyl pyrophosphate synthetase gene family inSaccharomycescerevisiae, is essential for cell viability in the absence of eitherPRS1 orPRS3 [J].J Bacteriol,1998,180(23):6 404-6 407.
[12]GIETZ R D,AKIO S. New yeast-Escherichiacolishuttle vectors constructed withinvitromutagenized yeast genes lacking six-base pair restriction sites [J].Gene,1988,74(2):527-534.
[13]J.萨姆布鲁克,D.W.拉塞尔.分子克隆实验指南(第三版)[M].黄培堂等译,北京:科学出版社,2002.
[14]GIETZ R D,SCHIESTL R H,WILLEMS A R,et al.Studies on the transformation of intact yeast cells by the LiAc / SS‐DNA/PEG procedure [J].Yeast,1995,11(4):355-360.
[15]F.M.奥斯伯,R.E.金斯顿,J.G.塞德曼等.新编分子生物学实验指南(第四版) [M].北京:科学出版社,2005.
[16]GONZALES K,KAYIKC O,SCHAEFFER D G,et al.Modeling mutant phenotypes and oscillatory dynamics in theSaccharomycescerevisiaecAMP-PKA pathway [J].BMC Systems Biology,2013,7(1):40.
Over-expressing key enzyme genes in the purine synthesis pathway by integrating into genome improves cyclic adenosine monophosphate production bySaccharomycescerevisiae
WANG Kai,JI Xiao-bing,XU Huan-huan,QIU Shen-shen,ZOU Shao-lan*
(College of Chemical Engineering, Tianjin University, Tianjin 300072, China)
Regulation of the expressing level of key enzyme genes in the purine synthesis pathway would influence cyclic adenosine monophosphate (cAMP) production by changing the metabolic flux to AMP, ADP and ATP. In this study, PRPP synthetase-encoding genePRS1 andPRS3, PRPP amidotransferase-encoding gene ADE4 and adenylate kinase-encodingADK1 were separately integrated into the genome of the cAMP-producing strain GA125 under the control of promoterPGK1pand terminatorPGK1tfrom phosphoglyceric kinase-encoding genePGK1. The integrating vector YIplac211 was used and cAMP production by the resultant strains was investigated by fermentation in shake flasks. It showed that when adenine was added into fermentation media, the cAMP titers at 120 h were (5 380.5±1.91) μmol/L, (5 189.4±1.72) μmol/L, (5 131.8±2.05) μmol/L and (5 199.9±1.62) μmol/L, respectively, and increased by 9.8%, 5.9%, 4.8% and 6.2%, respectively. On the other hand, over-expression ofADK1 gene led to a slight inhibition of growth. The adenine consumption of all strains was (0.399±0.01)~(0.447±0.02) g/L and it seemed that the glucose utilization and ethanol production didn’t vary with genes being over-expressed.
Saccharomycescerevisiae; purine synthesis pathway; integration; cyclic adenosine monophosphate; adenine
10.13995/j.cnki.11-1802/ts.201608005
硕士(邹少兰副研究员为通讯作者,E-mail:slzhou@tju.edu.cn)。
2015-12-17,改稿日期:2016-04-21
