基于XcmI识别序列的T载体的构建及连接PCR片段的优化*
2016-09-01张峄桥张雁芳龙超良李春悦龙学辉崔文玉
张峄桥, 张雁芳+, 龙超良, 李春悦, 龙学辉, 崔文玉, 张 浩△, 汪 海△
(1. 军事医学科学院卫生学环境医学研究所, 北京 100850; 2. 北京赛德维康药物研究院, 北京 100039)
基于XcmI识别序列的T载体的构建及连接PCR片段的优化*
张峄桥1, 张雁芳1+, 龙超良1, 李春悦1, 龙学辉1, 崔文玉2, 张浩1△, 汪海1△
(1. 军事医学科学院卫生学环境医学研究所, 北京 100850; 2. 北京赛德维康药物研究院, 北京 100039)
目的:构建基于XcmI识别序列的T载体并对与其连接的PCR片段进行优化。方法:首先将5’和3’端含有Xcm I识别序列的人组蛋白H4 cDNA定向克隆至pCDNA3.0表达载体中,再用Xcm I酶切得到基于pCDNA3.0载体骨架的T载体,为提高T载体与DNA片段的连接效率,在DNA片段PCR扩增前将其引物进行磷酸化并在PCR结束后再用Taq DNA聚合酶和dATP处理,分别将长度为312 bp和1 329 bp的PCR片段T载体连接并将连接产物转化大肠杆菌DH5α感受态细胞,培养转化子,通过菌液PCR和琼脂糖凝胶电泳估算转化子的阳性率。结果:经Xcm I酶切后的T载体能高效地与目的DNA片段连接;除T载体本身质量外,扩增DNA片段所用引物的磷酸化与否也是影响克隆效率的重要因素之一;对较大的插入片段而言,经PCR扩增、纯化后再用Taq DNA聚合酶和dATP处理也能够显著增加连接效率。结论:克服了对蓝白筛选或插入自杀基因等实验条件的限制,可为T载体的常规制备并与目的片段进行高效地连接提供了新的线索。
XcmI;T载体;磷酸化;dATP
分子克隆是基因功能研究和特定DNA片段的测序等实验中的必要环节,常规的分子克隆是通过具有特定识别序列的限制性内切酶分别对目的DNA片段和载体进行酶切之后再行连接、转化和阳性克隆的筛选。相对而言,T载体克隆是利用T载体3’端的dT和目的片段3’端的dA互补而进行的具有粘端特性的高效连接。由于该过程不依赖于限制性内切酶的切割作用,因而有很强的通用性,同时避免了酶切、回收等繁琐过程,因而更简单、经济。
现在制备T载体主要有以下两种方法:1是将目的载体用EcoR V酶切然后通过Taq DNA聚合酶或者末端核苷酸转移酶在其3’平端加入dT[1-3];2是在目的载体中先插入含有Xcm I或者Ahd I酶切位点的片段后再进行相应的酶切反应后得到3’含dT的片段[4-6]。第一种方法由于载体在酶切过程中发生不完全酶切的现象,极少量的未经酶切的载体会夹杂在T载体中而产生假阳性,同时一些载体没有类似EcoR V等能产生DNA平端的限制性内切酶而无法应用。此外,用Taq DNA聚合酶或者末端核苷酸转移酶在载体3’平端加入dT的效率也是个问题;第二种方法相对而言更简便,现在基于XcmI酶切构建T载体的研究比较多,其基本原理是插入含有两个Xcm I识别序列的DNA片段,在这两个识别序列中可以不包含或包含长度不一的DNA序列。同时其T载体骨架中往往含有蓝白筛选标记基因,通过蓝白筛选以及PCR筛选获得阳性克隆。本研究将5’和3’端含有Xcm I识别序列的人组蛋白H4 cDNA定向克隆至pcDNA3.0表达载体中,用Xcm I酶切后得到基于pcDNA3.0载体骨架的T载体,为检测T载体的质量,分别将长度为312 bp和1 329 bp的cDNA片段与载体连接并转化大肠杆菌DH5α感受态细胞,通过菌液PCR和琼脂糖凝胶电泳计算转化克隆的阳性率,同时对连接反应的条件进行了优化。
1 材料与方法
1.1材料与试剂
1.1.1材料大肠杆菌DH5α感受态细胞购自Takara公司,pcDNA3.0真核表达载体, pcDNA3.0-H4表达质粒(含有编码人组蛋白H4 312 bp cDNA片段), pcDNA3-CRBN表达质粒(含有编码人CRBN 1 329 bp的cDNA片段)为本室保存。
1.1.2主要试剂Taq DNA聚合酶、限制性内切酶Xcm I购自NEB公司;限制性内切酶BamH I、EcoR I、T4 多核苷酸激酶、T4 DNA连接酶、dATP、Premix Taq DNA聚合酶、DNA 2000 marker均为Takara公司产品,琼脂糖凝胶回收试剂盒、PCR产物回收试剂盒购自北京天根生物科技有限公司。
1.2方法
1.2.1引物设计及合成(1) 用于构建T载体的引物:正向引物:5’-CGCGGATCCCCA AAG CTC GAG TGG ATGTCTGGGCGAGGTAAAG-3’;反向引物:5’- CCGGAATTCCCA GAT ATC GAT TGG TCAACCGCCGAAACCATAAA-3’,其中斜体部分的碱基分别为限制性内切酶BamH I和EcoR I识别位点,划线部分的碱基为Xcm I限制性内切酶识别位点。(2) 用于扩增人H4 cDNA(312 bp)的正向引物:5’-CGGAATTCGCCACCATGTCTGGGCGAGGTAAAG-3’;反向引物序列为:5’-CGGGATCCACCGCCGAAACCATAAA-3’。(3) 用于扩增人CRBN cDNA(1329 bp)的正向引物:5’- CCCAAGCTTATGGCCGGCGAAGGAGATC-3’;反向引物:5’-CGGGATCC TTACAAGCAAAGTAT-TACTTTGTC-3’。(4) 用于菌液PCR鉴定阳性克隆的一对引物分别为:T7引物:5’-TAATACGACTCACTATAGG-3’; SP6引物: 5’-ATTTAGGTGACACTATAGAA-3’。以上引物均由上海生物工程有限公司合成,PAGE纯化。
1.2.2PCR引物的磷酸化及PCR扩增反应的条件在50 μl反应体系中分别加入2 μl dNTP (10 mmol/L), 5 μl primer (10 μmmol/L), 1 μl T4 多核苷酸激酶, 2 μl 10×T4多核苷酸激酶反应缓冲液, 10 μl灭菌的超纯水,37℃ 反应1 h。50 μl PCR 反应体系包含以下组分:1 μl 正向引物(10 μmmol/L),1 μl 反向引物(10 μmmol/L),1 μl dNTP(10mmmol/L),1 μl 质粒模板(pcDNA3.0-H4,0.01 μg/μl),5 μl Taq DNA buffer,0.5 μl Taq DNA聚合酶,40.5 μl 灭菌的超纯水。PCR扩增人组蛋白H4 cDNA反应条件为:95℃ 预变性2 min;95℃ 10 s,58℃ 30 s,72℃ 30 s,30 个循环;72℃ 延伸5 min。PCR扩增人CRBN cDNA的模板为(pcDNA3.0-CRBN,0.01 μg/μl),反应条件除了在30个循环中72℃ 90 s之外其余条件均相同,将PCR产物进行1%琼脂糖凝胶中电泳后切胶回收,加入50 μl回收。
1.2.3菌液PCR鉴定转化子的阳性率从转化平板中分别挑取10个克隆于500 μl LB中培养6 h左右待培养液变混浊,取1 μl至24 μl含相应引物的Premix PCR反应体系中,PCR反应条件与1.2.2相同,PCR反应结束后,取6 μl于1%琼脂糖凝胶中电泳,根据阳性条带出现的机率估算转化子的阳性率。
1.2.4基于Xcm I识别序列和pCDNA3骨架的T载体的构建取1 μg pCDNA3载体用BamH I+EcoR I双酶切,37℃反应5 h后进行0.7%琼脂糖凝胶中电泳后切胶回收,加入50 μl回收;将含Xcm I识别序列的正反向引物用pcDNA3.0-H4为模板扩增后用琼脂糖凝胶回收试剂盒回收300 bp左右的特异片段,将pCDNA3载体酶切片段和PCR片段按摩尔比1∶3的比例进行连接、转化,菌液PCR鉴定阳性克隆,随机挑取阳性克隆,提取质粒分别用BamH I+EcoR I双酶切和Xcm I单酶切分析切出的片段大小,鉴定正确的质粒进一步用T7引物进行序列分析确证,该质粒命名为pCDNA3-XcmI-H4-XcmI,将该质粒用Xcm I酶切后回收5.4 kb大小的片段即为T载体。
1.2.5T载体和PCR片段连接的条件优化将T载体用Xcm I酶切后用琼脂糖凝胶回收试剂盒回收大小为5.4 kb的片段,与用下列不同方案获得片段按摩尔比1∶3的比例进行连接、转化,菌液PCR鉴定阳性克隆、扩增。PCR扩增人组蛋白H4 cDNA分别用一对磷酸化和非磷酸化引物;PCR扩增人CRBN cDNA分别用一对磷酸化和非磷酸化引物,同时设一组在用磷酸化扩增、纯化后再用Taq酶处理,反应体系为:35 μl纯化的PCR产物,5 μl ThermoPol 缓冲液, 0.5 μl Taq DNA聚合酶,2 μl dATP (10 mmol/L)和7.5 μl灭菌的纯水,72℃反应。
2 结果
2.1pCDNA3-XcmI-H4-XcmI质粒及T载体的构建
将5’和3’端分别含有BamH I、Xcm I以及Xcm I、EcoR I识别位点的人组蛋白H4 cDNA片段用BamH I+EcoR I双酶切后与用同样双酶切处理的pCDNA3片段连接转化后,菌液PCR初步鉴定阳性克隆,提取质粒,在分别用BamH I+EcoR I双酶切和Xcm I单酶切鉴定(图1),空载体pCDNA3用BamH I+EcoR I双酶切后仅有5.4 kb片段大小的单一条带(图1,第2泳道),表明pCDNA3载体含有BamH I或EcoR I识别位点且这两个位点之间没有大的片段,而pCDNA3-XcmI-H4-XcmI质粒用这两个酶消化后得到了5.4 kb大小的片段以及300 bp左右大小的片段(图1,第3泳道),表明人组蛋白H4 cDNA片段已经插入至pCDNA3载体的BamH I和EcoR I位点之间; pCDNA3载体用Xcm I处理并电泳后其DNA带型与未经酶切的pCDNA3载体一致(图1,第4泳道),说明在pCDNA3载体中没有Xcm I识别位点,而pCDNA3-XcmI-H4-XcmI质粒用Xcm I处理后得到5.4 kb大小的片段以及300 bp左右大小的片段(图1,第5泳道),表明该质粒引入了两个Xcm I位点,且在这两个位点之间含有300 bp的片段,结果与预期一致。将该质粒用T7引物测序后,序列完全正确。将pcDNA3-XcmI-H4-XcmI质粒用Xcm I酶切后回收5.4 kb即为T载体,其DNA双链两端的5’端均含有突出的“T”。
2.2T载体与人组蛋白H4 cDNA PCR片段的连接及其条件的优化
在实验初期,用未磷酸化的引物扩增H4 cDNA片段并与T载体按摩尔比3∶1比例连接,转化后挑取10个转化子培养,取1 μl菌液用T7+SP6引物进行PCR和琼脂糖凝胶电泳鉴定,结果见图2A,在10个PCR产物中有6个出现阳性条带,表明此时阳性率仅为60%,为进一步提高连接效率,同时考虑到PCR产物的5’端如果具有磷酸基团则可以与T载体3’端T的羟基连接从而增强连接效率。因此在PCR扩增前先将引物进行磷酸化,在扩增并与T载体连接,菌液PCR后经琼脂糖凝胶电泳,结果见图2B,在10个PCR产物均出现阳性条带,表明此时阳性率达到100%。将阳性转化子转接至LB培养基中培养,提取质粒后用T7引物进行测序,结果表明插入的片段为人组蛋白H4 cDNA,且在靠近T7引物序列的接头处含有“T”(图3A),在靠近SP6引物序列的接头处含有“A”(图3B)。
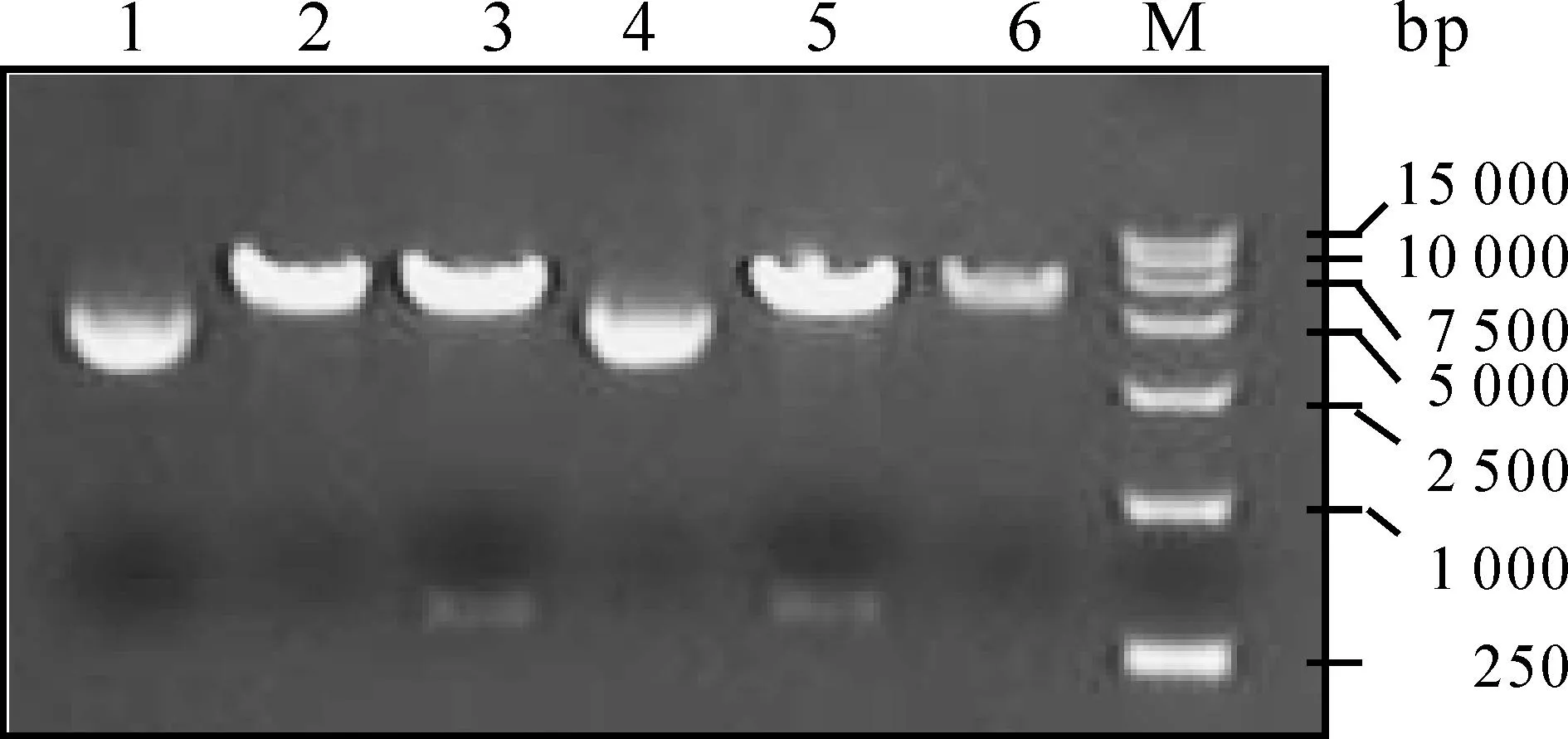
Fig. 1Restriction endonuclease analylsis of recombinant pcDNA3-XcmI-H4-XcmI plasmids
1: pcDNA3-XcmI-H4-XcmI plasmids without any restriction endonuclease digestion; 2: pcDNA3.0 plasmid digested with BamH I+EcoR I; 3: pcDNA3-XcmI-H4-XcmI plasmid digested with BamH I + EcoR I; 4: pcDNA3.0 plasmid digested with Xcm I; 5: pcDNA3-XcmI-H4-XcmI plasmid digested with Xcm I; 6: purified T vector plasmid; M: 2 000 bp DNA marker
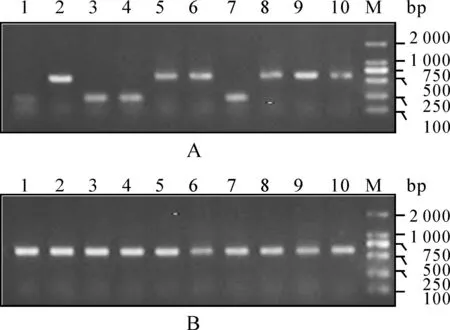
Fig. 2Determination of the positive rates of the transformants in both of the non-phosphorylated group
A: Phosphorylated group; B: By routine PCR with T7+SP6 primer pair set; Lane 1-10: Amplification of H4 cDNA with the templates of cultured bacteria transformed with the ligation solution containing T-vector and the amplified coding sequence of human histone H4 cDNA; M: 2000 bp DNA marker
2.3T载体与人CRBN cDNA PCR片段的连接及其条件的优化
考虑到H4 cDNA PCR片段较小,仅为300 bp左右,较容易连接至载体,我们进一步扩增较大长度的DNA片段并将之与T载体连接。实验中以本实验室的pCDNA3-CRBN为模板扩增人CRBN cDNA片段(长度为1.3 kb左右),首先比较了引物未发生磷酸化及磷酸化对连接效率的影响,未磷酸化组的转化子中,10个PCR产物中仅有2个为阳性(图4A),其阳性率为20%,而磷酸化组的10个随机挑选的转化子中有5个出现阳性的1.4 kb大小的条带,其阳性率为50%(图4B),虽然和未磷酸化组相比有显著提高,但是阳性率仍然不高。为此,将磷酸化组的PCR片段回收后用Taq酶和dATP进一步处理,将处理并纯化后的产物再与T载体按摩尔比3∶1比例连接,转化后用菌液PCR进行鉴定,结果见图4C,可以看出随机挑选的10个转化子中有9个出现长度约为1.4 kb大小的片段,其阳性率达到90%,可基本满足普通实验的需要。将阳性转化子转接至LB培养基中培养,提取质粒后用T7和SP6引物进行测序,结果表明插入的片段为CRBN cDNA,且在靠近T7引物序列的接头处含有T(图5A),在靠近SP6序列的接头处含有“A”(图5B),(用SP6反向引物测序时,序列与原序列互补,故用方框标记的是“T”)。
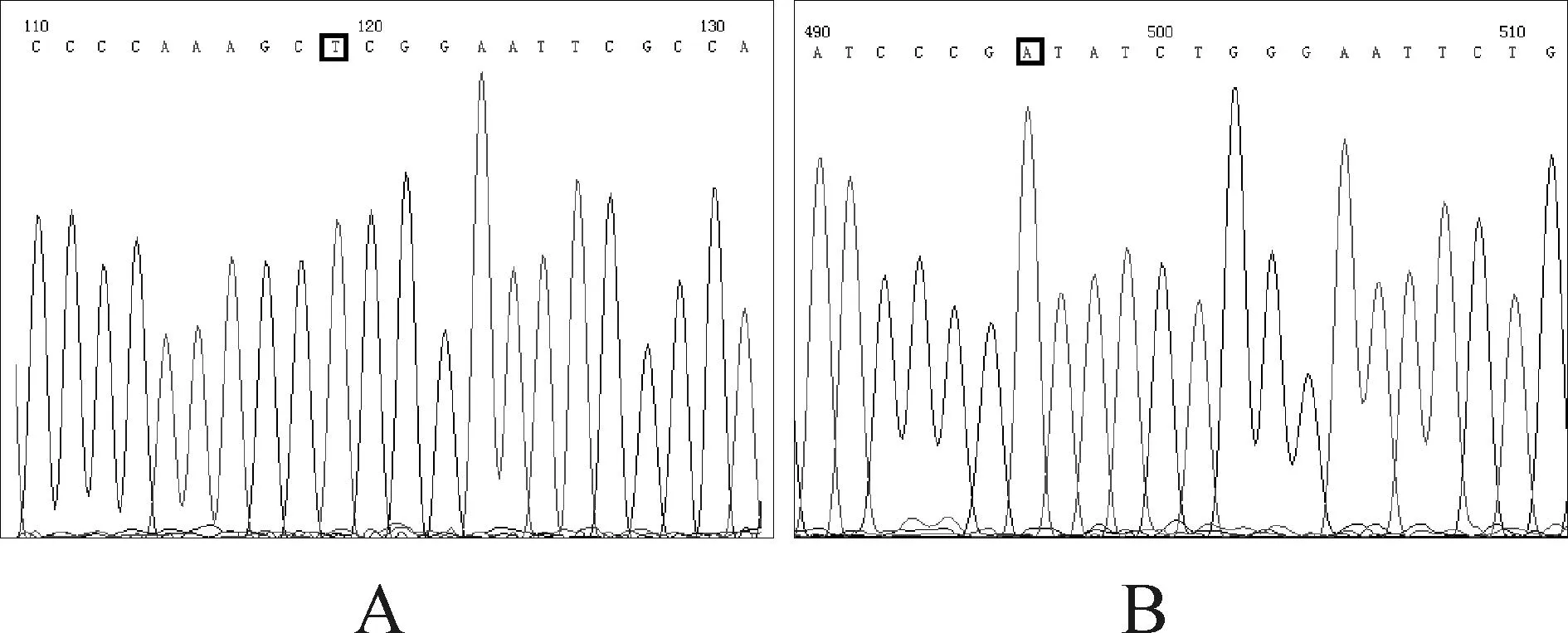
Fig. 3DNA sequencing results of the plasmid extracted from transformants of the ligation mixture containing T-vector and the amplified coding sequence of human histone H4 cDNA by T7 primer showing the appearance of “T”
(A) near the 5’ end of the junction and “A” in the 3’ end of the junction (B)
3 讨论
T载体克隆由于不受限制性内切酶识别序列的限制且操作简单、经济而广泛应用于各种用途的分子克隆实验中。比如经特定实验筛选出的差异DNA片段的测序(甲基化片段、特定抗体的染色质免疫共沉淀片段等)、融合蛋白在原核或真核细胞中的表达、基因敲低、启动子大片段的克隆、抗体库的构建等[7-10],在如何制备T载体的问题上已有广泛和深入的报道[11,12]。但在如何优化T载体与插入片段的连接条件问题上的研究相对较少。但是这个问题很重要,因为最终连接的效率不仅仅取决于制备的T载体本身的质量,还与待连接片段的质量相关。
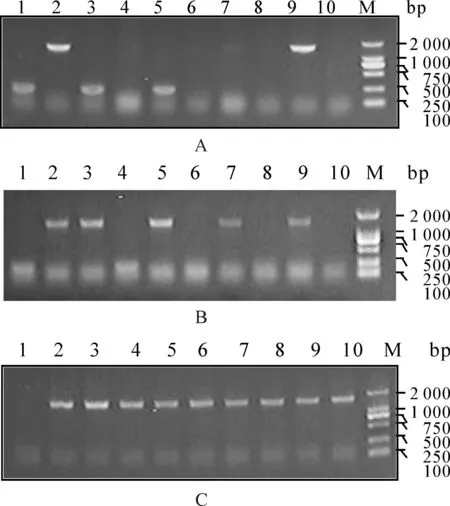
Fig. 4Determination of the positive rates of the transformants in non-phosphorylated group
A: Phosphorylated group; B: Phosphorylated plus retreatment with Taq group; C: By routine PCR with T7+SP6 primer pair set; Lane 1-10: Amplification of CRBN cDNA with the templates of cultured bacteria transformed with the ligation solution containing T-vector and the amplified coding sequence of human CRBN cDNA; M: 2 000 bp DNA marker
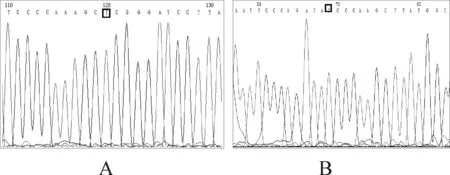
Fig. 5DNA sequencing results of the plasmid extracted from transformants of the ligation mixture containing T-vector and the amplified coding sequence of human CRBN cDNA by T7 and SP6 primer showing the appearance of “T” (A) near the 5’ end of the junction and “A” in the 3’ end of the junction (B)
有关T载体的制备已有很多报道。在本实验中我们首先在pCDNA3载体中插入了两端含Xcm I识别序列的人组蛋白H4 cDNA 片段,得到pCDNA3-XcmI-H4-XcmI质粒,该质粒用Xcm I酶切之后即可得到载体双链DNA两端的3’端均含T的片段和H4 cDNA 片段,进一步回收大片段即为T载体。该方案的优点在于用Xcm I处理pCDNA3-XcmI-H4-XcmI质粒制备T载体时,经酶切后的T载体片段与少量的未发生酶切的pCDNA3-XcmI-H4-XcmI质粒相差300 bp,经0.7%琼脂糖凝胶电泳后可以分开从而避免了原始质粒的污染;此外通过引物设计,只要该质粒的两个Xcm I位点被切割,切割后的DNA片段的两端就能够产生突出的T,如果只有一个位点发生切割,由于其片段较大而再琼脂糖凝胶电泳时会与T载体分离,这些都保证了T载体的质量。
本研究通过几个对照实验逐步发现了影响T载体与DNA插入片段连接效率的重要因素。在插入人组蛋白H4 cDNA时,用普通未磷酸化的引物进行扩增时,发现T载体与其连接效率仅为50%,后来又对连接比例、感受态的类别进行了变换,发现均不能提高连接效率(结果未显示),考虑到T载体的3’端为羟基,而用未磷酸化引物扩增的DNA片段的5’端也为羟基,T4DNA连接酶不能将二者连接,因此,在PCR扩增前首先将正反向引物的5’端分别磷酸化,此时PCR扩增后的片段在与T载体连接后转化子的阳性率达到100%。在将长度为1 300 bp的PCR片段与T载体连接时,同样发现引物经磷酸化后其PCR扩增片段与T载体的连接效率由20%提高到50%,说明引物的磷酸化确实是影响连接效率的重要因素之一。但是50%的连接效率还是太低,考虑到在PCR过程中Taq DNA聚合酶虽然在每轮PCR结束后可优先在PCR产物的3’端加入一个“A”,但是随着DNA片段长度的增加,其在PCR产物的3’端加入“A”的效率会逐渐降低。因此,PCR产物经纯化后需再用Taq DNA聚合酶和dATP对其处理以提高其3’端加入“A”的效率。实验结果表明,经该步处理后,连接效率由50%提高至90%,表明该步骤在将长片段DNA PCR扩增产物连接至T载体的过程中发挥重要作用,这也提示可以将待扩增的DNA片段先用高保真的DNA聚合酶扩增,而后再用普通的Taq DNA聚合酶和dATP对其处理以高效地和T载体连接。这样既保证了DNA序列的正确同时又有利于连接的高效率,在分子克隆实验中将非常有用。
为增加T载体与DNA片段后筛选出的几率,大多实验通过蓝白筛选实验进行初筛。一方面使实验过程变得复杂,同时该实验依赖于蓝白筛选标记基因--LacZ基因的存在,对于不含该基因的目标载体则无法进行T载体的克隆,而且即便是白色的克隆,其是否为阳性仍需要进行PCR或测序进一步验证[13-15]。本实验通过对连接反应条件的优化表明要增加T载体与DNA插入片段的连接效率,一方面需要高质量的T载体,同时插入片段DNA两端5’的磷酸化与否及其3’端突出A的比例对连接效率发挥重要作用。如果这些条件都得到优化,可以达到不低于90%的连接效率。
[1]Zhou MY, Gomez-Sanchez CE. Universal TA Cloning[J].CurrIssuesMolBiol, 2000, 2(1): 1-7.
[2]Marchuk D, Drumm M, Saulino A,etal. Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products[J].NucleicAcidsRes, 1990, 19(5): 1154.
[3]Holton TA, Graham MW. A simple and efficient method for direct cloning of PCR products using ddT-tailed vectors[J].NucleicAcidsRes, 1990, 19(5): 1156.
[4]Gu J, Ye C. pYEMF, a pUC18-derived XcmI T-vector for efficient cloning of PCR products[J].MolBiotechnol, 2011, 47(3): 229-233.
[5]You L, Luo J, Wang A,etal. A hybrid promoter-containing vector for direct cloning and enhanced expression of PCR-amplified ORFs in mammalian cells[J].MolBiolRep, 2010, 37(6): 2757-2765.
[6]Jeung JU, Cho SK, Shim KS,etal. Construction of two pGEM-7Zf(+) phagemid T-tail vectors using AhdI-restriction endonuclease sites for direct cloning of PCR products[J].Plasmid, 2002, 48(2): 160-163.
[7]卢银平, 郭涛, 祝建芳, 等. 通用表达型T载体的构建与真核基因的快速克隆表达[J]. 实用医学杂志, 2009, 25(22): 3740-3742.
[8]Lv Y, Wang T, Yuan S,etal. T-vector and in vivo recombination as tools to construct a large antibody library of breast cancer[J].Hybridoma(Larchmt), 2010, 29(3): 251-254.
[9]Luo K, Harding SA, Tsai C. A modified T-vector for simplified assembly of hairpin RNAi constructs[J].BiotechnolLett, 2008, 30(7): 1271-1274.
[10]Goda N, Tenno T, Takasu H,etal. The PRESAT-vector: asymmetric T-vector for high-throughput screening of soluble protein domains for structural proteomics[J].ProteinSci, 2004, 13(3): 652-658.
[11]Jo C, Jo SA. A simple method to construct T-vectors using XcmI cassettes amplified by nonspecific PCR[J].Plasmid, 2001, 45(1): 37-40.
[12]Zhao Y, Liu Z, Yu S,etal. Construction of a High Efficiency PCR Products Cloning T Vector Using pGEM-5zf (+). Avicenna[J].AvicennaJMedBiotechnol, 2009, 1(1): 37-39.
[13]Shimada T, Yamamoto K, Ishihama A. Novel Members of the Cra Regulon Involved in Carbon Metabolism in Escherichia coli[J].JBacteriol, 2011, 193(3): 649-659.
[14]Walrad PB, Hang SY, Gergen JP. Hairless is a cofactor for Runt-dependent transcriptional regulation[J].MolBiolCell, 2011, 22(8): 1364-1374.
[15]Li ZF, Blissard GW. Cellular VPS4 Is Required for Efficient Entry and Egress of Budded Virions of Autographa californica Multiple Nucleopolyhedrovirus[J].JVirol, 2012, 86(1): 459-472.
Construction of T vectors based on Xcm I recognition site and optimization of PCR fragments for ligation
ZHANG Yi-qiao1, ZHANG Yan-fang1+, LONG Chao-liang1, LI Chun-yue1, LONG Xue-hui1,CUI Wen-yu2, ZHANG Hao1△, WANG Hai1△
(1. Institute of Health and Environmental Medicine, Academy of Military Medical Sciences, Beijing 100850;2. Beijing Thadweik Academy of Medicine, Beijing 100039, China)
Objective: To construct T vectors based on Xcm I recognition site and optimize the PCR fragments for its ligation. Methods: We firstly cloned the human histone H4 cDNA containing one Xcm I recognition site at both its 5’ and 3’ end into pCDNA 3.0 vector and then generated T vector with pCDNA 3.0 backbone by cutting the recombinant plasmid with Xcm I. To increase the ligation efficiency, the primers were firstly phosphorylated before DNA fragments amplification and then the PCR products were treated with Taq DNA polymerase and dATP after PCR amplification. Two DNA fragments with the length of 312 bp and 1 329 bp were ligated to it and the ligation mixture was transformed into E.coli DH5α competent cells and the positive rates of the transformants were evaluated by PCR and DNA agarose gel electrophoresis. Results: Our results showed that the T vector produced by our method could ligate to the target DNA fragments with high efficiency. Besides, the phosphorylation state of the primers used for PCR amplification is also an important factor determining the cloning efficiency. What’s more, as for longer DNA fragments, retreatment with Taq DNA polymerase and dATP after PCR amplification and purification could improve the ligation efficiency significantly. Conclusion: Our protocol may overcome the dependence on blue/white screening to get positive clones and provide a potent way to generate T vectors and ligate them to the target PCR fragment.
Xcm I, T vector, phosphorylation, dATP
国家重点基础研究发展计划资助(973计划)(2012CB518200);+:共同第一作者
2015-03-31
2015-10-21
△Tel:010-66932651; E-mail: wh9588@sina.com, zhanghal197@hotmail.com;?+:共同第一作者
Q786
A
1000-6834(2016)01-046-05
10.13459/j.cnki.cjap.2016.01.012
