P38MAPK在低氧及炎症联合刺激诱导人肺微血管内皮细胞凋亡中的作用
2016-08-22吉圣珺魏晓群莫冠文梅湛强张培芳
吉圣珺,魏晓群,莫冠文,梅湛强,张培芳
·论著·
P38MAPK在低氧及炎症联合刺激诱导人肺微血管内皮细胞凋亡中的作用
吉圣珺1,魏晓群1,莫冠文1,梅湛强1,张培芳1
目的 探讨P38MAPK在低氧及炎症联合刺激诱导人肺微血管内皮细胞凋亡中的作用。方法 将人肺微血管内皮细胞株进行复苏及传代培养,分为空白对照组、低氧组(低氧6 h、12 h、24 h)、肿瘤坏死因子(TNF-α)刺激组(用10 ng/ml、20 ng/ml、50 ng/ml、100 ng/ml的TNF-α进行刺激)、模型组(低氧24 h+TNF-α100 ng/ml联合刺激)及通路阻断剂组(加入通路阻断剂SB203580进行干预)。Western-blot法分析各组细胞中P38丝裂原活化蛋白激酶(P38MAPK)表达,流式细胞术分析各组细胞半胱氨酸天冬氨酸蛋白酶-3(Caspase-3)活力,流式细胞术和TUNEL法联合检测细胞凋亡。结果 与空白对照组相比,模型组p-P38MAPK表达升高(P<0.05)。与低氧组(24 h)、TNF-α刺激组(100 ng/ ml)相比,模型组(低氧24 h+TNF-α100ng/ml联合刺激)细胞活力均下降(P均<0.01);与模型组相比,通路阻断剂组细胞Caspase-3活力下降(P<0.01)。与模型组相比,通路阻断剂组细胞凋亡率及凋亡指数均下降(P<0.01)。结论 P38MAPK在低氧及炎症联合刺激诱导的人肺微血管内皮细胞中具有促凋亡作用,P38MAPK 通路阻断剂对低氧及炎症联合刺激损伤的人肺微血管内皮细胞具有保护作用。
P38MAPK;人肺微血管内皮细胞;细胞凋亡
慢性阻塞性肺疾病(COPD)是常见的严重危害人类健康的呼吸系统疾病。肺动脉高压(PH)作为其主要并发症之一,被认为是影响COPD预后的独立危险因素[1],而其发生机制又与肺动脉内皮细胞的凋亡密切相关[2]。在COPD患者中,由于持续存在的缺氧及炎症状态导致肺血管内皮细胞的凋亡增加,出现了肺动脉的重构,最终导致严重的PH。丝裂原活化蛋白激酶(MAPK)是一类广泛存在于哺 乳动物细胞内的丝氨酸/苏氨酸蛋白激酶,是细胞内重要的信号转导通路,参与细胞的生长、分化、凋亡等许多生理过程[3]。目前有关于MAPK参与内皮细胞凋亡研究最多的信号通路包括:P38MAPK信号通路、ERK通路等。本研究拟通过检测P38MAPK通路在低氧及肿瘤坏死因子(TNF-α)联合刺激诱导人肺微血管内皮细胞(HPMVECs)凋亡模型中的活化情况,探讨P38MAPK在缺氧及炎症联合刺激诱导人肺微血管内皮细胞凋亡中的作用。
1 材料与方法
1.1实验材料 人肺微血管内皮细胞株购于上海细胞库,10%胎牛血清及高糖细胞培养基购于美国HyClone公司,四甲基偶氮唑蓝购于美国Sigma公司,Caspase-3活性检测试剂盒购于美国的BD公司,HRP羊抗兔武汉BOSTER公司,Phospho-p38 MAPK购自美国Cell Signaling Technology公司,GAPDH购自北京康为世纪生物科技有限公司,P38MAPK阻断剂(SB203580)购自美国Sigma公司,流式细胞仪购于美国的BD公司,光学显微镜购于重庆奥特光学仪器有限公司,电热恒温培养箱购于上海博迅实业公司KPL公司,AnnexinV /PI凋亡检测试剂盒购于瑞士Roche公司,蛋白印迹实验设备购自美国 BioRad 公司。
1.2方法
1.2.1HPMVEC的复苏和传代培养 从液氮保存罐中取出冻存管(含原代HPMVEC株),立即放入37℃水浴中,快速摇晃,直至冻存液完全融化。将其置于含10%胎牛血清的DMEM培养液中,在37℃ 、5%二氧化碳培养箱中培养,隔天换液。内皮细胞呈鹅卵石样贴壁生长,当细胞生长至90%融合时用0.25%乙二胺四乙酸(EDTA)胰蛋白酶进行消化传代,取第四代血管内皮细胞进行实验。
1.2.2实验分组及处理 分为空白对照组(用10% 胎牛血清的 DMEM 培养基处理内皮细胞,置于体积分数5%、气体成分为二氧化碳培养箱中常规培养),低氧刺激组(低氧6 h、12 h、24 h,用10%胎牛血清的DMEM培养基处理内皮细胞,置于气体成分为体积分数3%氧气、5%二氧化碳和 92%氮气的三气培养箱中进行低氧培养),TNF-α刺激组(用10 ng/ml、20 ng/ml、50 ng/ml、100 ng/ml的TNF-α进行刺激,然后用10%胎牛血清的 DMEM 培养基处理内皮细胞,置于体积分数5%、气体成分为二氧化碳培养箱中常规培养)、模型组(低氧24 h+TNF-α100 ng/ml)、通路阻断剂组(在模型组中加入通路阻断剂SB203580进行干预)。
1.2.3流式细胞术检测半胱氨酸天冬氨酸蛋白酶-3(Caspase-3)活力 分别收集各组细胞,1500 r/min,离心10 min,预冷的PBS洗涤细胞两次,300 g,4℃离心5 min,收集5×105细胞;加TF2-DEVD-FMK 37℃反应1 h;流式细胞计分析caspase-3阳性细胞数和平均荧光强度。
1.2.4流式细胞术和TUNEL法联合检测细胞凋亡每组取3个6孔板的培养孔,胰酶消化法(不含EDTA)收集细胞,1000 g离心5 min,弃上清,PBS液重悬细胞后,离心,重复2次后弃上清。分别加入BindingBuffer液200重悬并进行细胞计数,调整细胞数至105个/ml。取200 μl细胞悬液,加入5 μl Annexin-FITC混匀,室温避光10 min。再加入5 μl PI染液混匀,流式细胞仪进行检测。流式细胞仪分析:每份样品取1×104个细胞PI染色,做DNA分析。TUNEL法检测:采用原位末端标记法(TUNEL)检测各组HPMVEC凋亡情况,并计算凋亡指数(AI):待测细胞用多聚甲醛固定,Triton-100柠檬酸钠处理,每孔加入45 μl TUNEL标记液,5 μl末端转移酶溶液,37℃温育2 h,冲洗,用荧光显微镜观察,用DAB显色的标本加入50 μl TUNEL显色液,37℃温育30 min,DAB室温显色10 min,PBS洗涤3次×5 min,苏木素复染数秒,流水冲洗,梯度酒精脱水,晾干,中性树胶封片,光学显微镜下观察棕黄色的细胞核为阳性结果,蓝色为苏木素复染的细胞核。
1.2.5统计学处理 采用SPSS13.0统计学软件进行分析,计量资料以均数±标准差(±s)表示,多组比较采用单因素方差分析,组间对比采用Tukey法分析,P<0.05为差异有统计学意义。
2 结果
2.1MTT比色法检测的细胞活力 低氧6 h、12 h、空白对照组的相对细胞活力无差异,但显示有逐步下降的趋势,而低氧24 h组的相对细胞活力明显低于空白对照组(P=0.041)(表1)。TNF-α10 ng/ml、20 ng/ml、50 ng/ml分别与TNF-α100 ng/ml组间存在统计学差异(P <0.05),而TNF-α100ng/ml的相对细胞活力明显低于空白对照组(P=0.006),TNF-α对细胞活力的抑制与TNF-α浓度呈现剂量依赖关系(表2)。与低氧24 h、TNF-α100 ng/ml分别单独作用组比较,模型组(低氧24 h+TNF-α100 ng/ ml联合刺激)的相对细胞活力更低,存在统计学差异(P值分别为0.002、0.005)(表3)。
表1 不同低氧刺激时间下HPMVEC的活力(±s)
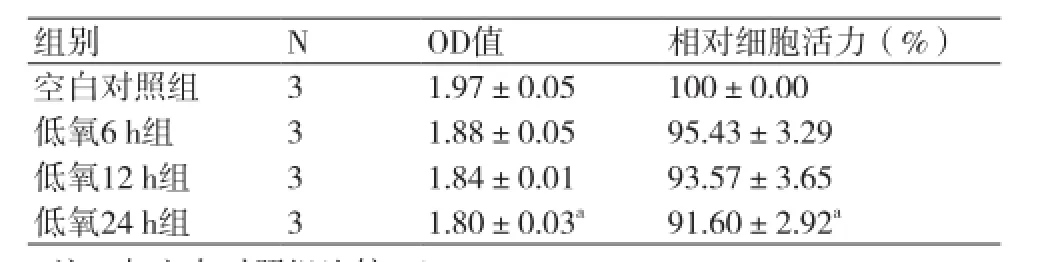
表1 不同低氧刺激时间下HPMVEC的活力(±s)
注:与空白对照组比较,aP<0.05
组别NOD值相对细胞活力(%)空白对照组31.97±0.05100±0.00低氧6 h组31.88±0.0595.43±3.29低氧12 h组31.84±0.0193.57±3.65低氧24 h组31.80±0.03a91.60±2.92a
表2 不同TNF-α浓度刺激下HPMVEC的活力(±s)

表2 不同TNF-α浓度刺激下HPMVEC的活力(±s)
注:与空白对照组比较,aP<0.05;与TNF-α10 ng/ml比较,bP <0.05;与TNF-α20 ng/ml 比较,cP<0.05;与TNF-α50 ng/ml比较,dP<0.05
组别NOD值相对细胞活力(%)空白对照组31.97±0.05100±0.00 TNF-α10 ng/ml组31.84±0.0193.68±3.30 TNF-α20 ng/ml组31.81±0.02a92.24±2.55aTNF-α50 ng/ml组31.71±0.02a87.30±2.64aTNF-α100 ng/ml组31.53±0.02abcd77.93±2.97abcd
2.2Western blot检测p-P38MAPK 与空白对照组相比,模型组p-P38MAPK表达升高(1.75± 0.25),存在统计学差异(P=0.035),根据上述磷酸化蛋白表达情况,加入P38MAPK通路阻断剂SB203580后,可使p-P38MAPK的表达下降(0.92±0.25),与模型组比较,存在统计学差异(P=0.015)(图1)。
2.3Caspase-3的活力检测 通路阻断剂组(16.89%±0.76%)与模型组(25.39%±0.72%)相比,caspase-3的活性明显下降,存在统计学差异(P=0.001)(表4)。
表3 各实验组刺激下HPMVEC的活力(±s)

表3 各实验组刺激下HPMVEC的活力(±s)
注:与空白对照组比较,aP<0.05;与低氧24 h组比较,bP <0.05;与TNF-α100ng/ml组比较,cP<0.05
组别N OD值相对细胞活力(%)空白对照组31.97±0.05100±0.00低氧24 h组31.80±0.03a91.60±2.92aTNF-α100 ng/ml组31.53±0.02ab77.93±2.97ab低氧24 h+TNF-α100 ng/ml组31.30±0.01abc65.93±1.87abc
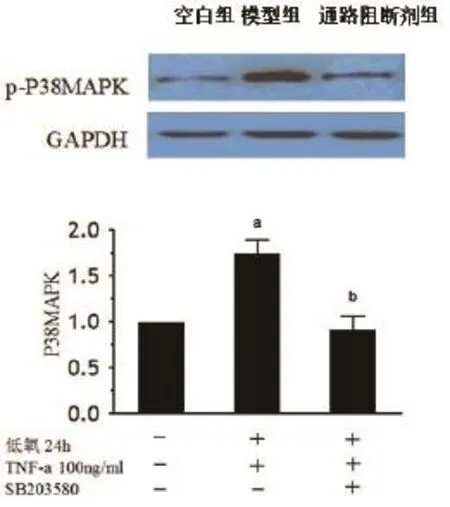
图1 各实验组p-P38MAPK的比较[与空白对照组比较,aP<0.05;与模型组(低氧24 h+TNF-α100 ng/ml组)比较,bP<0.05]
2.4流式细胞仪鉴定细胞凋亡 通路阻断剂组(16.45%±0.10%)与模型组(25.53%±0.94%)相比,细胞凋亡率明显下降,存在统计学差异(P<0.001)(图2)。
2.5TUNEL检测细胞凋亡 与模型组(86.00% ±1.06%)相比,通路阻断剂组(37.13%± 1.01%)细胞凋亡指数下降,存在统计学差异(P <0.001)(图3)。
3 讨论
表4 各实验组细胞Caspase-3的活力比较(±s)

表4 各实验组细胞Caspase-3的活力比较(±s)
注:与空白对照组比较,aP<0.05;与模型组(低氧24 h+TNF-α100ng/ml组)比较,bP<0.05
组别NCaspase-3的活力(%)空白对照组30.39%±0.22%低氧24 h+TNF-α100 ng/ml组325.39%±0.72%aSB203580+低氧+TNF-α组316.89%±0.76%ab

图2 各实验组细胞凋亡率的比较[与空白对照组比较,aP<0.05;模型组(低氧24 h+TNF-α100 ng/ml组)比较,bP<0.05]
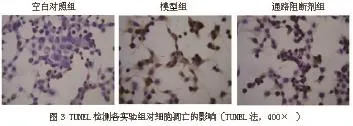
图3 各实验组细胞凋亡率的比较[与空白对照组比较,aP<0.05;模型组(低氧24 h+TNF-α100 ng/ml组)比较,bP<0.05]
COPD引起的PH具有多因性,缺氧及炎症引起肺血管收缩及重构是COPD发生PH的重要病理生理基础。研究发现,血管内皮细胞凋亡是PH发病的重要机制[4,5]。缺氧或炎症(高浓度的TNF-α)均可通过诱导血管内皮细胞的凋亡,诱导肺动脉血管壁重构,导致PH的发生[6-8]。然而,目前许多关于COPD相关性PH的体外细胞模型的建立均以缺氧或炎症因子作为单一诱导因素,COPD患者却往往同时存在的缺氧及炎症的复合状态。因此,COPD相关的PH体外细胞模型,在缺氧及炎症两者复合因素刺激下建立,更符合疾病的病理生理状态。而抗血管内皮细胞凋亡的研究,将对延缓COPD患者PH的发展具有重要的意义。
MAPK信号通路在血管内皮细胞凋亡过程中发挥着及其重要的作用,但由于其可被多种上游信号激活,激活后又可导致多种转录因子的表达,并与其他信号通路存在相互作用。因此,P38MAPK可因刺激物和底物的不同,在血管内皮细胞凋亡过程中发挥不同的效应[9]。有研究者在缺氧复氧诱导大鼠肺内皮细胞凋亡的模型中,证实了激活的P38MAPK发挥抗凋亡作用,而加入P38MAPK阻断剂 SB203580后则导致缺氧复氧的内皮细胞凋亡增加[10]。本研究发现,低氧及TNF-α联合刺激HPMVEC可导致磷酸化P38MAPK表达升高,同时伴有Caspase-3活力升高,而加入P38MAPK 抑制剂(SB203580)后,可显著降低人肺微血管内皮细胞的凋亡,这一结果与李泉[11]等在CD8+T淋巴细胞诱导血管内皮细胞凋亡的检测结果一致。
本研究采用Annexin V-FITC/PI双染法及TUNEL法检测低氧及TNF-α联合刺激对HPMVEC凋亡的影响,结果显示低氧及TNF-α联合刺激细胞凋亡率及凋亡指数较正常对照组明显增加,而加入P38MAPK通路阻断剂(SB203580)后,可显著降低细胞凋亡率及凋亡指数。本研究结果证实了低氧及炎症联合刺激诱导人肺微血管内皮细胞的凋亡与P38MAPK的磷酸化激活密切相关,P38MAPK通路阻断剂(SB203580)可抗低氧及炎症联合刺激诱导的人肺微血管内皮细胞凋亡。P38MAPK通路在缺氧联合炎症刺激人肺微血管内皮细胞凋亡中具有促凋亡作用,该研究结果有望为COPD继发PH的防治提供新的思路和靶点。
[2] Vallerie V,McLaughlin,Stephen L,et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians;American Thoracic Society, Inc; and the Pulmonary Hypertension Association[J]. J Am Coll Cardiol,2009,53(17):1573-619.
[3] 李德冠,樊飞跃,孟爱民. P38MAPK通路在造血系统调节中的作用[J]. 中国药理学通报,2011,27(1):4-6.
[4] Winn RK,Harlan JM. The role of endothelial cell apoptosis in inflammatory and immune diseases[J]. 2005,3(8):1815-24.
[5] Sage E,Mercier O,Van den Eyden F. Endothelial cell apoptosis in chronically obstructed and reperfused pulmonary artery[J/OL]. Respiratory Research,2008,12(2):9-19.
[6] Taraseviciene·Stewart L,KasaharaY,Alger L,et al. Inhibition of the VEGF Receptor 2 combined with chronic hypoxia causes cell deathdependent pulmonary endothelial cell proliferation and severe pulmonary hypertension[J]. 2001,15(2):427-38.
[7] Zhao YD,Campbell AI,Robb M. Protective role of angiopoietin-1 in experimental pulmonary hypertension[J]. Circulation Research,2003,92(9):984-91.
[8] 金惠铭,刘清行,张国平,等. TNF-α引起的微血管内皮细胞功能障碍及其分子机制[J]. 2000,16(10):940-1.
[9] Thompson CB. Apoptosis in the pathogenesis and treatment of disease[J]. Science,1995,267(5203):1456-62.
[10] Wang X,Zhou YS,Kim HP,et al. Hepatocyte growth factor protects against hypoxia /reoxygenation induced Apoptosis In endothelial cells[J]. 2004,279(7):5237-43.
[11] 李泉,张剑,黎纬明,等. 异基因CD8+T淋巴细胞诱导血管内皮细胞凋亡的分子机制[J]. 中华医学杂志,2009,89(24):1693-7.
本文编辑:芦洁,田国祥
Role of P38MAPK in human pulmonary microvascular endothelial cells apoptosis induced by hypoxia and inflammation combination
JI Sheng-jun*, WEI Xiao-qun, MO Guan-wen, MEI Zhan-qiang, ZHANG Pei- fang. *The first people's Hospital of Foshan. Foshan, 528000, China.
ZHANG Pei-fang, E-mail: zhangpf0757@163.com
Objective To investigate the effect of p38mitogen-actvated protein kinase (P38MAPK) on the apoptosis of human pulmonary microvascular endothelial cells induced by hypoxia and inflammation. Methods Human pulmonary microvascular endothelial cell line was recovered, subcultured, divided into blank control group,hypoxia group (hypoxia 6 h, 12 h, 24 h) and TNF alpha stimulation group (10 ng/ml, 20 ng/ml, 50 ng/ml, 100 ng/ml TNF alpha), model group (hypoxia combined with 100 ng/ml TNF alpha ) and pathway blocker group (SB203580). P38MAPK expression in each group was detected by Western blotting. Caspase-3 activity in each group was analyzed by flow cytometry. Apoptosis were detected by combination of flow cytometry and TUNEL assay. Results Compared with the blank control group, p-P38MAPK expression of model group was increased (P<0.05). Caspase-3 activity of model group (24 h hypoxia combined with 100ng/ml TNF alpha) was reduced compared with hypoxia group (24 h) and TNF alpha stimulation group (100 ng/ml) (P<0.01); Caspase-3 activity of pathway blocker group was reduced compared with model group (P<0.01). Apoptosis rate and apoptotic index were reduced in pathway blocker group than model group (P<0.01). Conclusion P38MAPK plays a role in promoting in human pulmonary microvascular endothelial cells apoptosis induced by hypoxia and inflammation. P38MAPK pathway blocker (SB203580) has protective effect on hypoxia and inflammation in human pulmonary microvascular endothelial cells.
P38MAPK; Human pulmonary microvascular endothelial cells; Apoptosis
R563
A
1674-4055(2016)06-0676-04
佛山市卫生及计生局医学科研立项课题(2015255)
1528000 佛山,广东省佛山市第一人民医院呼吸科
张培芳,E-mail:zhangpf0757@163.com
10.3969/j.issn.1674-4055.2016.06.09
[1] M,Nakano K,Hiramoto T,et al. Significance of pulmonary artery pressure in emphysema patients with mild-to-moderate hypoxemia[J]. Respir Med,2003,97(8):915-20.
