夏枯草H PLC融合指纹图谱研究
2016-08-04李敏陈蕾周倩王亮郭威
李敏 陈蕾 周倩 王亮 郭威
(1济南市食品药品检验检测中心,山东 济南250102;2深圳福田区中医院,广东 深圳518034;3山东中医药大学药学院,济南 250355;4山东省中医药研究院,济南 250014)
夏枯草H PLC融合指纹图谱研究
李敏1陈蕾2周倩3,4王亮4郭威3,4
(1济南市食品药品检验检测中心,山东 济南250102;2深圳福田区中医院,广东 深圳518034;3山东中医药大学药学院,济南 250355;4山东省中医药研究院,济南 250014)
目的:建立夏枯草的融合指纹图谱,实现同分异构体熊果酸和齐墩果酸完全色谱分离和同步含量测定,对夏枯草质量进行评价。方法:采用高效液相色谱法,建立夏枯草的指纹图谱,并为互为同分异构体的熊果酸和齐墩果酸单独建立分析方法,使其达到完全色谱分离,使用“MATLAB R2012a版”将两个色谱图融合部分进行基线融合计算,使用“中药色谱指纹图谱相似度评价系统2004A版”进行图谱融合。结果:建立了可单独、分段操作的“模块式”融合指纹图谱,共标定了18个共有色谱峰,结合质谱和对照品对部分共有峰进行了推断,其中1,3~ 8,17,18号峰分别推测为丹参素、咖啡酸、芦丁、金丝桃苷、异槲皮苷、异迷迭香酸苷、迷迭香酸、齐墩果酸和熊果酸。同时对15批夏枯草样品中熊果酸和齐墩果酸的含量进行了测定,熊果酸含量0.19%~0.36%,齐墩果酸含量0.055%~ 0.11%。结论:建立的熊果酸和齐墩果酸的分析方法分离度高、分析速度快、结果准确可靠;建立的“模块式”融合指纹图谱可以更准确全面反映夏枯草中的化学成分,可为夏枯草质量控制研究提供参考。
夏枯草;融合指纹图谱;熊果酸;齐墩果酸;同分异构体;高效液相色谱
夏枯草是临床常见中药,为唇形科植物夏枯草(Prunella vulgaris L.)的干燥果穗,有清肝泻火,明目,散结消肿的功效[1]。现代药理学研究表明,夏枯草的主要活性化学成分为三萜类、甾体类、黄酮类、香豆素类[2],其中熊果酸和齐墩果酸分别属于乌苏烷型和齐墩果烷型五环三萜,具有保肝、抗炎、抗病毒、抗微生物、抗糖尿病、抗肿瘤等作用,是夏枯草的主要有效成分[3-4],也是夏枯草质量评价的重要含量测定指标,但是由于它们之间除1个甲基的取代位置略有不同外,化学结构完全相同,因此很难分离[5-8]。指纹图谱由于其可全面反映药物中化学成分的种类和数量而作为重要的质量评价方法得到广泛的应用,但其研究的侧重点在于短时间内反映大量成分的信息,因此即使指纹图谱分析时间相对较长,结构相近的同分异构体同样很难得到理想的分离效果。
目前融合指纹图谱的研究多为不同检测手段图谱的融合[8],或因所含成分紫外吸收波长不一致而将不同波长的图谱融合[9],基于同分异构体的定量分析方法建立融合指纹图谱的研究未见报道。为更加准确、真实地鉴定夏枯草的质量,本文拟采用高效液相色谱法(HPLC)建立夏枯草的指纹图谱,并针对其中难以分离的熊果酸和齐墩果酸单独建立特征分析方法,使用“MATLAB R2012a版”结合“中药色谱指纹图谱相似度评价系统2004A版”实现两个图谱的融合,以建立准确反映夏枯草化学成分的融合指纹图谱,同时对其中熊果酸和齐墩果酸两种同分异构体的含量进行测定,以实现对夏枯草质量更加客观和准确的评价。
1仪器与材料
1.1仪器
1200系列高效液相色谱系统 (美国安捷伦公司),BP211D型电子天平(德国赛多利斯),Simplicity纯水仪(美国密理博公司),LC-350A超声波中药处理机(济宁市中区鲁超仪器厂)。
1.2材料
熊果酸对照品(批号:X-006-140801-2),齐墩果酸对照品(批号:Q-003-140731-1),均购自成都瑞芬思科技生物有限公司,乙腈为色谱纯,水为超纯水,磷酸等其他试剂均为分析纯,夏枯草为市售中药饮片,经山东省中医药研究院鉴定为唇形科植物夏枯草的干燥果穗。
2方法与结果
2.1对照品溶液的制备
取熊果酸和齐墩果酸对照品适量,精密称定质量,分别加甲醇制成每1 mL含熊果酸0.201 0 mg,含齐墩果酸0.180 0 mg的溶液,作为对照品储备溶液。
2.2供试品溶液的制备
取各供试品粉末(过2号筛)0.25 g,精密称定,精密加入甲醇25 mL,称重,超声提取30 min,冷却至室温,用甲醇补足减失的质量,摇匀,滤过,取续滤液过0.45 μm微孔滤膜,即得。
2.3色谱条件
2.3.1夏枯草指纹图谱色谱条件Agilent Eclipse XDB-C18(250 mm ×4.6 mm,5 μm)色谱柱;以乙腈(A)-0.1%磷酸水(B)为流动相;梯度洗脱(0~10 min,5% ~ 18%A;10~ 35 min,18%A;35~40 min,18%~30%A;40~55 min,30%A;55~ 75 min,30% ~ 100%A;75~ 100 min,100%A);检测波长 210 nm;流速1.0 mL/min;柱温30℃;进样量10 μL。色谱图见图1。
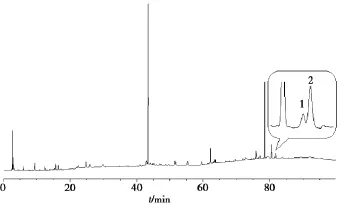
图1 夏枯草指纹图谱
2.3.2熊果酸和齐墩果酸色谱条件Agilent E-clipse XDB-C18(250 mm ×4.6 mm,5 μm)色谱柱;以乙腈-0.1%氨水(87∶13)为流动相;检测波长210 nm;流速 1.0 mL/min;柱温 30℃;进样量5 μL。色谱图见图2。
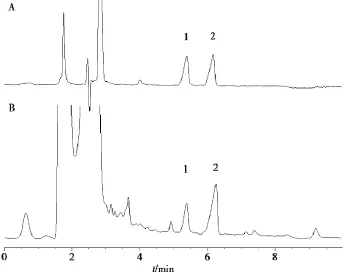
图2夏枯草含量测定HPLC图
2.4融合指纹图谱的建立
使用“Chemstation B.01.03版”工作站将“2.3.1”和“2.3.2”中的色谱图导出为AIA文件,使用“中药色谱指纹图谱相似度评价系统 2004A版”将AIA转换为TXT格式,抽取两个色谱图中熊果酸和齐墩果酸的色谱峰数据,使用“MATLAB R2012a版”将两个色谱图中熊果酸和齐墩果酸的数据进行基线融合计算,将“2.3.1”中指纹图谱中的对应数据替换为融合计算后的数据,得到的TXT文件导入“中药色谱指纹图谱相似度评价系统2004A版”得到融合指纹图谱,见图3。
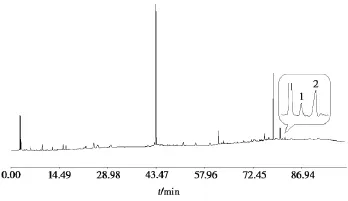
图3 夏枯草融合指纹图谱
2.5方法学考察
2.5.1精密度试验取夏枯草样品的供试品溶液,分别按照“2.3.1”和“2.3.2”中色谱条件连续测定6次,按照“2.4”中方法将色谱图融合,以熊果酸的保留时间和峰面积为参照,分别对各共有峰相对保留时间和相对峰面积进行统计。结果表明,各共有色谱峰的相对保留时间和相对峰面积基本一致,RSD均 <3%,表明仪器精密度良好。
2.5.2稳定性试验取夏枯草供试品溶液在室温下保存,于 0,3,6,12,24 h分别按照“2.3.1”和“2.3.2”中色谱条件测定,按照“2.4”中方法将色谱图融合,以熊果酸保留时间和峰面积为参照,分别对各共有峰相对保留时间和相对峰面积进行统计。结果表明,各共有色谱峰的相对保留时间和相对峰面积基本一致,RSD均 <3%,表明样品在24 h内稳定性良好。
2.5.3重复性试验取夏枯草样品(过2号筛)6份,按照“2.2”中方法制备供试品溶液,分别按照“2.3.1”和“2.3.2”中色谱条件依次测定,按照“2.4”中方法将色谱图融合,以熊果酸保留时间和峰面积为参照,分别对各共有峰相对保留时间和相对峰面积进行统计。结果表明,各共有色谱峰的相对保留时间和相对峰面积基本一致,RSD<3%,表明方法重复性良好。
2.5.4熊果酸和齐墩果酸线性和线性范围考察精密吸取混合对照品储备液,分别稀释至储备液浓度的0.025,0.05,0.1,0.3,0.5倍,制成熊果酸和齐墩果酸的系列标准溶液,按照“2.3.2”色谱条件进行测定,测定峰面积,分别以峰面积积分值(y)对对照品浓度(x,mg/mL)进行线性回归处理,熊果酸的回归方程为:
y=2 360.7x+4.95,
相关系数r为0.999 9,线性范围为0.005 0~0.100 5 mg/mL;齐墩果酸的回归方程为:
y=3 306.9x+2.158 3,
相关系数r为0.999 9,线性范围为0.004 5~0.090 0 mg/mL。
2.5.5熊果酸和齐墩果酸加样回收试验取夏枯草样品(过2号筛)6份,分别按样品含量-对照品(1∶1)的大致比例加入一定量的对照品溶液,按照“2.2”中方法制备供试品溶液,按照“2.3.2”中方法进行测定,计算回收率。结果测得熊果酸和齐墩果酸的回收率平均值分别为 97.16%和 98.72%,RSD分别为1.68%和 1.01%,表明方法测定结果准确,见表1。
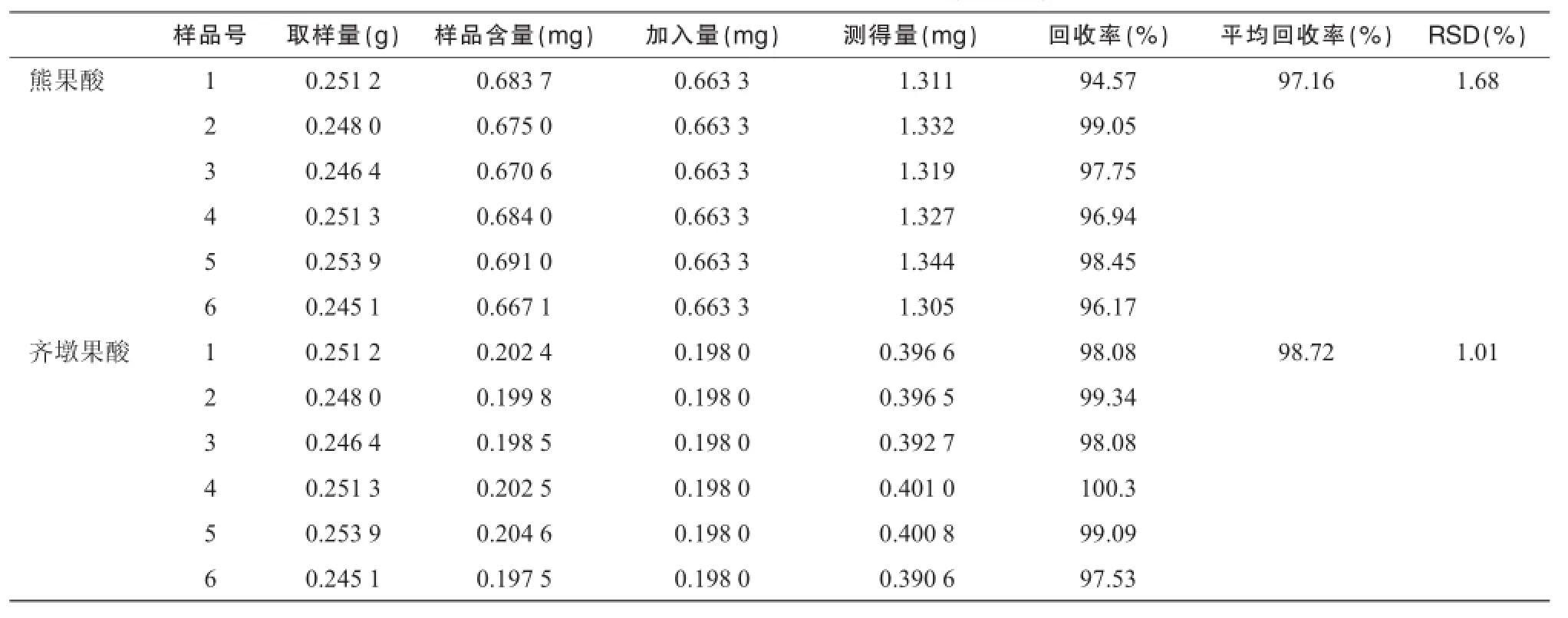
表1 夏枯草饮片回收率试验结果(n=6)
2.6夏枯草饮片融合指纹图谱共有模式的建立
将按照“2.4”中方法所得15批夏枯草融合指纹图谱以TXT格式依次导入“中药色谱指纹图谱相似度评价系统2004A版”软件。以1号夏枯草饮片色谱图作为参照图谱,经过多点校正、自动匹配,以中位数法,生成对照图谱,并标定了18个共有色谱峰。夏枯草饮片HPLC融合指纹图谱叠加图和对照图谱见图4。
使用“中药色谱指纹图谱相似度评价系统2004A版”进行相似度分析,15批次夏枯草与相应对照图谱的相似度均大于0.90,符合指纹图谱要求。
2.7指纹图谱质谱分析
按照“2.3.1”中色谱条件,将磷酸替换为0.3%甲酸,对夏枯草样品进行电喷雾多级串联质谱(ESI-MSn)分析,对共有峰进行了推测和推断。由正负模式的准分子离子峰可以推断1,4,5,6,7,8号峰的分子量分别为 198,610,464,464,522和360 m/z,根据保留时间并结合文献报道[10-12],推测1号峰为丹参素;结合对照品判断3号峰为咖啡酸;4号峰裂解方式与文献一致,推断为芦丁;5,6号峰裂解方式一致,一级质谱均产生162 m/z中性丢失,剩余部分裂解方式与文献报道中槲皮素一致,推断为槲皮素的六碳糖苷类,结合夏枯草成分报道和保留时间数据,推断5,6号峰分别为金丝桃苷和异槲皮苷;推断8号峰为迷迭香酸,7号峰发生162 m/z中性丢失后,剩余部分裂解方式与迷迭香酸相同,推断7号峰为异迷迭香酸苷;结合对照品判断 17,18号峰为齐墩果酸和熊果酸。见表2。

图4 夏枯草饮片HPLC融合指纹图谱叠加图和对照图谱
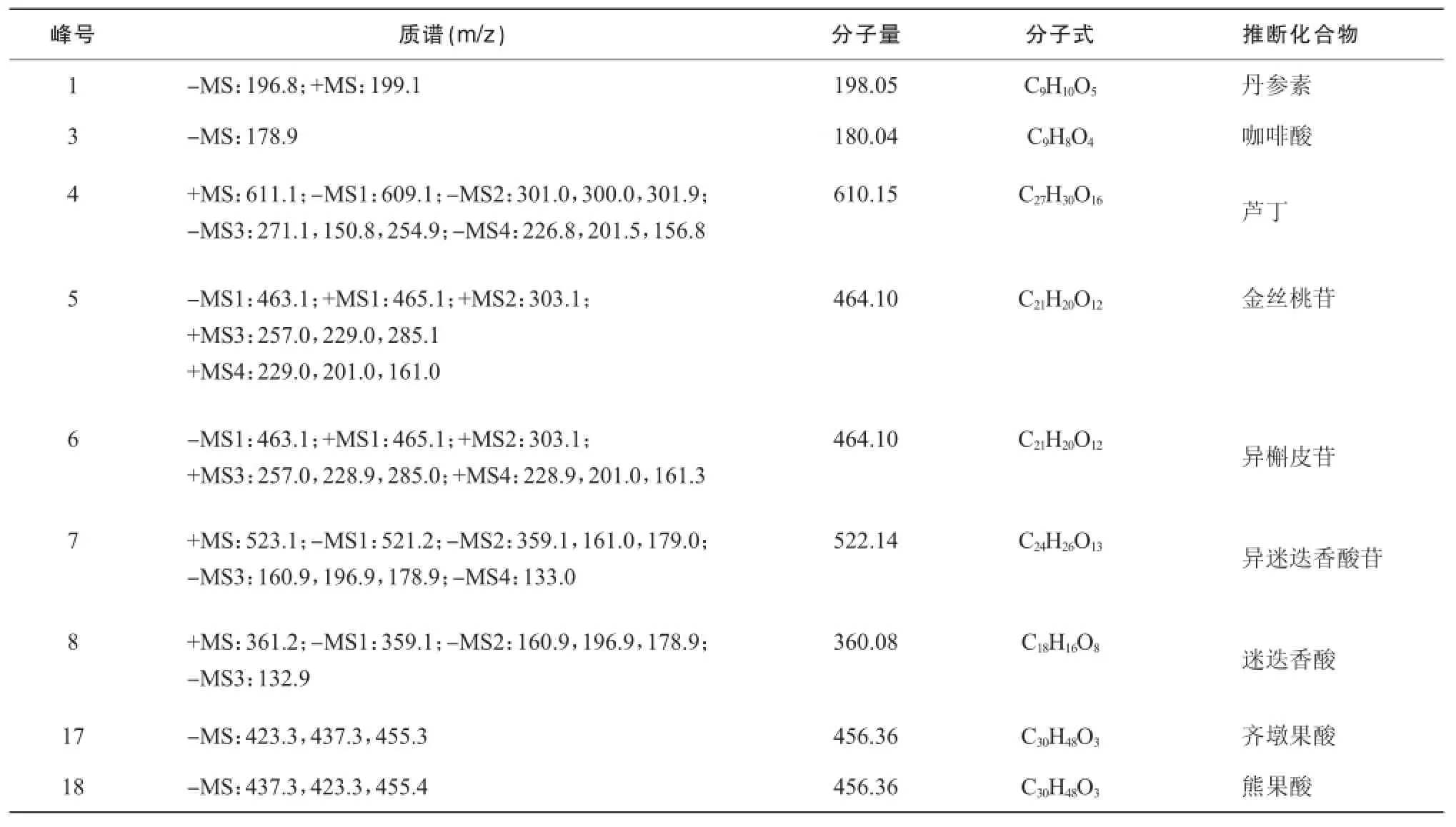
表2 夏枯草指纹图谱质谱分析
2.8主成分聚类分析
运用SPSS 19.0软件中的因子分析对共有峰进行标准化处理。以特征值>1为提取标准,前5个成分的特征值大于1,分别为6.588,4.374,2.117,1.389,1.139,累计方差贡献率达86.705%,能反映指纹图谱主要信息,因此提取前5个成分进行分析,计算主成分得分见表3。
由表 2中的主成分得分,运用SPSS 19.0软件对15个批次夏枯草样品采用组间平均数联结法,以欧式平方距离对样品聚类,得到聚类分析树状图,见图5。对样品分类进行判别分析得到区域图,见图6。
通过聚类分析可以将样品分为3类,样品1,3,5~ 13,15为一类;2,14为一类;4为一类。判别分析结果表明分类合理。从分析结果可以看出,夏枯草样品总体质量较为稳定,少数批次成分差别较大,分类结果没有明显的地域特征。
2.9熊果酸和齐墩果酸含量测定
取夏枯草供试品溶液,按照“2.3.2”色谱条件进行测定,测定峰面积,分别从标准曲线上计算出供试品溶液中熊果酸和齐墩果酸的浓度,计算样品含量,结果见表3。
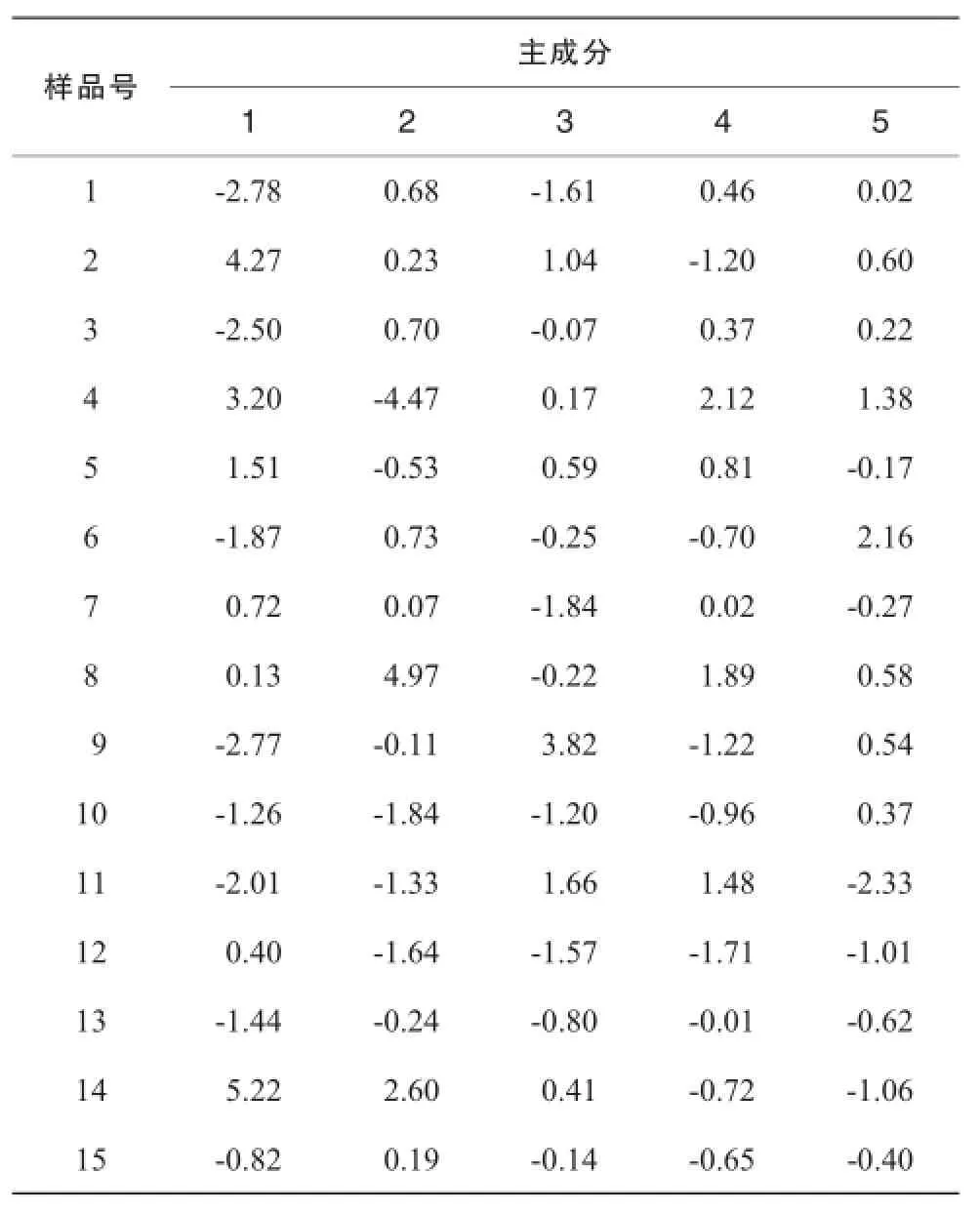
表3 夏枯草指纹图谱主成分分析
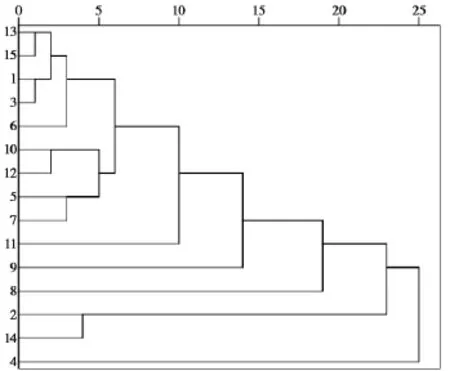
图5 夏枯草系统聚类树状图
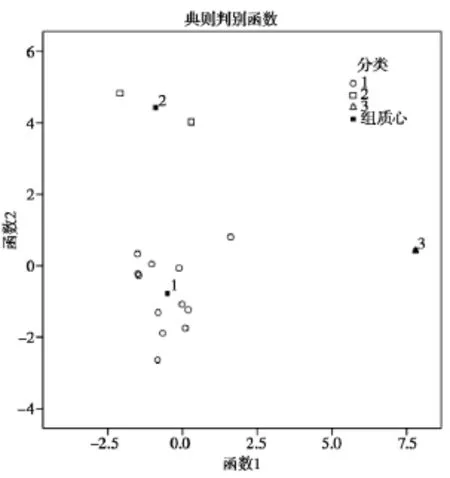
图6 夏枯草判别分析区域图
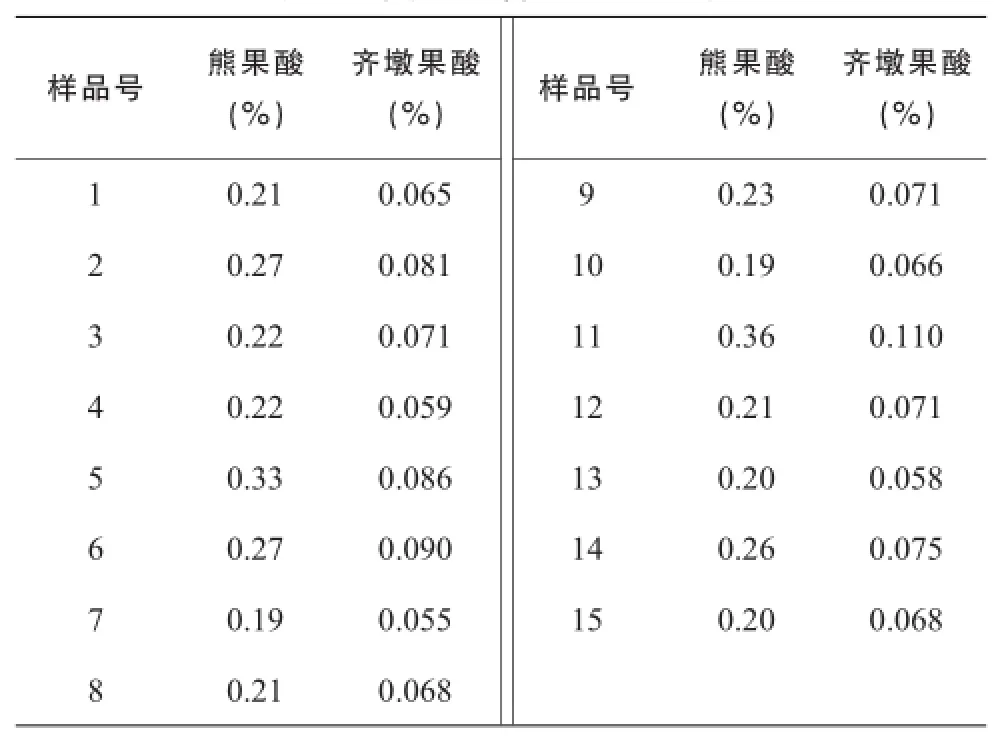
表4 夏枯草含量测定结果
3讨论
夏枯草中熊果酸和齐墩果酸均为五环三萜类化合物,除1个甲基的取代位置略有不同外,化学结构完全相同,因此分离一直是指纹图谱研究的难点。二者含量测定虽已有文献报道[5-8],但实验室中重现不理想,很难达到基线分离,分离时间也较长,为15~ 45 min;且由于报道中均采用甲醇组成流动相,熊果酸和齐墩果酸的检测波长为210 nm,接近甲醇的截止波长,导致基线不易平衡,信噪比很低。本研究中采用乙腈替代甲醇,很好地解决了信噪比低的问题;为改善二者分离度,同时在流动相中加入氨水,不足7 min即实现了两种同分异构体的基线分离,分离度达到 3.01。推测分离机理可能为,加入氨水后熊果酸和齐墩果酸中的羧基被离子化,成为强亲水性基团,消除了羧基在色谱柱C18填料上的保留,从而提高了熊果酸和齐墩果酸中非极性部分在色谱柱C18填料上吸附的选择性,进而实现了色谱分离。再通过与传统指纹图谱的融合,很好地实现了快速、高效、全面评价饮片质量的目的。
《中国药典》2015年版用迷迭香酸含量评价夏枯草质量,控制目标单一,不能反映夏枯草的其他有效成分。本研究建立了夏枯草饮片的融合指纹图谱,并同时对熊果酸和齐墩果酸进行含量测定,各色谱峰分离度较好,基线较平稳,色谱信息较为丰富,主要色谱峰相对保留时间基本一致,而且色谱峰分布均匀,15个批次夏枯草共标定了18个共有峰,可为夏枯草的质量控制提供参考。通过对比,不同批次夏枯草指纹图谱中共有峰相对保留时间较为一致,但相对峰面积有明显差别,测得熊果酸的含量在0.19%~ 0.36%之间,齐墩果酸的含量在0.055% ~ 0.11%之间,成分含量差别较大,因此仅控制迷迭香酸含量并不能反映夏枯草的真实质量,建议采用指纹图谱对夏枯草进行多成分同步质量控制。
本研究将熊果酸和齐墩果酸的定量分析图谱和夏枯草指纹图谱进行了融合,建立了“模块式”的融合指纹图谱,并结合质谱技术部分消除了单一测定方法指纹图谱的模糊性。在利用融合指纹图谱对药材进行评价时,除了可以更全面、准确地评价药材中的化学成分外,对融合成分可以直接选用相应的“模块”色谱条件快速、准确地进行测定,不再需要采集整个指纹图谱数据,从而大大节省分析时间,可以预见,随着研究的深入,融合指纹图谱中的“模块”越来越多,针对在实际应用中不同的质量控制要求会有越来越灵活高效的解决方案。融合指纹图谱的整体性好,信息量丰富,分离度更优化,可为真实、快速鉴别中药材质量提供参考,对于实际应用具有重要意义。
[1] 国家药典委员会.中华人民共和国药典(一部)[S].北京:中国医药科技出版社,2015:280.
[2] 窦景云,于俊生.夏枯草药理作用及临床应用研究进展[J].现代医药卫生,2013,29(7):1039-1041.
[3] 张明发,沈雅琴.齐墩果酸和熊果酸的抗微生物和原虫药理研究进展[J].抗感染药学,2010,7(3):153-156.
[4] 刘伟,丁海杰.HPLC测定夏枯草中熊果酸、齐墩果酸、迷迭香酸的含量[J].中成药,2008,30(4):577-580.
[5] 邸学,王海波,翟延君,等.HPLC测定藤梨根中熊果酸、齐墩果酸的含量[J].中国实验方剂学杂志,2012,18(1):66-68.
[6] 张兰珍,巴寅颖,季思伟,等.RP-HPLC测定夏枯草不同部位熊果酸和齐墩果酸含量[J].药物分析杂志,2009,29(9):1547-1549.
[7] 姚静.HPLC测定不同产地泽兰药材中齐墩果酸和熊果酸含量[J].中国实验方剂学杂志,2015,21(10):80-82.
[8] 李全斌,何开勇.齐墩果酸含量测定的研究进展[J].中国执业药师,2011,8(7):32-34.
[9] 崔媛,王小明,杨勇,等.薏苡仁油融合指纹图谱研究[J].中草药,2014,45(12):1698-1701.
[10] 方芳,王晶,方舟,等.复方黄白胶囊的多波长融合HPLC指纹图谱[J].中国实验方剂学杂志,2013,19(17):113-117.
[11] 梁杰康,张琳,严晓明.HPLC-ESI-MS/MS鉴定夏枯草的主要化学成分[J].中国中医药现代远程教育,2013,11(14):153-154.
[12] 封亮,贾晓斌,陈彦,等.夏枯草化学成分及抗肿瘤机制研究进展[J].中华中医药杂志,2008,23(5):428-434.
[13] 袁丽春,刘斌,石任兵.HPLC法测定不同市售荷叶药材中金丝桃苷和异槲皮苷的含量[J].药物分析杂志,2010,30(1):41-44.
Study on Fusion Fingerprint of Prunellae Spica by HPLC
Li Min1,Chen Lei2,Zhou Qian3,4,Wang Liang4,Guo Wei3,4
(1 Jinan Center for Food and Drug Control,Shandong Jinan 250102,China;2 Shenzhen Futian Hospital of TCM,Guangdong Shenzhen 518034;3 School of Pharmaceutical Sciences,Shandong University of Traditional Chinese Medicine,Jinan 250355;4 Shandong Academy of Traditional Chinese Medicine,Jinan 250014)
Objective:To establish the fusion fingerprint of prunellae spica,perform the complete chromatographic separation and synchronous content determination of two isomers,ursolic acid and oleanolic acid,and evaluate the quality of prunella spica.Methods:The fingerprint of prunellae spica was established by HPLC,and a special assay for ursolic acid and oleanolic was established for complete chromatographic separation.The baselines of the fusion part in the two chromatograms were calculated and corrected by“MATLAB R2012a”,and the chromatograms were fused by“Similarity Evaluation System for Chromatographic Fingerprint of TCM 2004A”.Results:A“modular”fusion fingerprint which could be performed individually and partly was established,in which18 common peaks were included.Thepeaks 1,3~ 8,17 and 18 were characterized by MS and standard samples,which were danshensu,caffeic acid,rutinum,hyperoside,isoquercitrin,salviaflaside,rosmarinic acid,oleanolic acid and ursolic acid,respectively.The determination results of ursolic acid and oleanolic acid contents in 15 batches of prunellae spica were 0.19%~0.36%and 0.055%~0.11%respectively.Conclusion:The developed method for determination of ursolic acid and oleanolic acid showed a high separation resolution,which was rapid,accurate and reliable.The established“modular”fusion fingerprint reflected the chemical composition of prunellae spica comprehensively and accurately,which provided a reference for the study on quality control of prunella spica.
Prunellae Spica;Fusion Fingerprint;Ursolic Acid;Oleanolic Acid;Isomers;HPLC
10.3969/j.issn.1672-5433.2016.04.006
深圳市福田区卫生公益性科研项目(FTWS2014028);山东省中医药科技发展计划(2015-167)
李敏,女,工程师。研究方向:中药检验和中药质量控制。E-mail:568027183@qq.com周倩,女,硕士,助理研究员。主要从事中药炮制研究。通讯作者E-mail:merveilleqq@163.com
2012-12-25)
