17α羟化酶缺乏症(46,XX)病例讨论
2016-07-05孙爱军北京市顺义区妇幼保健院北京0300北京协和医院北京0003
程 炜,孙爱军(. 北京市顺义区妇幼保健院,北京 0300; .北京协和医院,北京 0003)
·病例分析·
17α羟化酶缺乏症(46,XX)病例讨论
程 炜1,孙爱军2
(1. 北京市顺义区妇幼保健院,北京101300;2 .北京协和医院,北京100032)
1 临床资料
姓名:任××,出生日期:1989年12月14日,病案号:1507745
第一次就诊,2006年11月14日,17岁,内分泌科。
主诉:发现低血钾、高血压3年。
现病史:近3年发现低血钾,最低1.35 mmol/L,间断静脉补钾及口服补钾。同时发现血压高,平时160/100 mmHg,最高血压170/140 mmHg。在当地医院住院治疗,可疑诊断为特发性醛固酮增多症或17α羟化酶缺乏症。
既往史:否认。
月经史:原发闭经。
家族史:有一妹,正常。否认家族中有类似病史。
查体:身高:160 cm,体重:41 kg;BP:160/100 mmHg,HR:60次/min;心肺未及异常,肝脾未及;乳腺Ⅰ~Ⅱ级(追问患者曾用过倍美力,具体不详);女性幼稚型外阴,无阴毛;双下肢无水肿。
染色体:46,XX。
CT:双侧肾上腺增生。
诊断:17α羟化酶缺乏症?
处理:三全;血总皮质醇、ACTH;24 h尿UFC;立位PRA、AT-Ⅱ、Ald;FSH、LH、E2、T;复查染色体;盆腔超声。
第2次就诊,2006年12月5日,17岁,内分泌科。
K:2.90 mmol/L↓;肝肾功大致正常。
性激素:FSH:93.1 IU/L↑,LH:43.5 IU/L↑,
E2:6.24 pmol/L↓,T:0 ng/mL↓。
卧立位醛固酮试验:Ald卧位9.0毫微克/L↑,立位10.0毫微克/L;PRA卧位0.2,立位0.2↓;AT-Ⅱ卧位124.1↑,立位115.2↑。
ACTH:95.2 pg/mL↑。
血总皮质醇:0.24 μg/dL↓。
24小时尿游离皮质醇(UFC):<0.20 μg/24hr↓
染色体:46,XX。
超声:膀胱后方见子宫样结构,大小2.0×0.8×0.7 cm,肌层回声均,隐约可见内膜强回声;左附件区见卵巢样结构1.7×0.8 cm;右附件区件卵巢样结构1.8×0.9 cm。提示:幼稚子宫,卵巢发育不良?。
诊断:17α羟化酶缺乏症。
处理:地塞米松0.75 mg,1次/d;2~3月后可减量为0.375 mg,1次/d;建议妇科内分泌科就诊。
当日就诊于妇泌科。
病史同前。原发闭经,无第二性征发育,FSH↑,LH↑,E2↓,T↓,高血压,低血钾,ACTH↑,染色体:46,XX。
查体:乳房Ⅰ~Ⅱ级,无阴毛腋毛,有阴道。
诊断:17α羟化酶缺乏症(46,XX)。
处理:克龄蒙1片,1次/d,口服;利维爱1.25 mg,1次/d,口服;钙片600 mg,1次/d,口服。3个月后复查B超。
1、高血压、低血钾的常见原因有哪些?
2、原发闭经的常见原因有哪些?
3、17α羟化酶缺乏症发病机制?
4、17α羟化酶缺乏症有哪些特征及其原理?
5、可疑17α羟化酶缺乏症需要作哪些临床检查?
6、性激素化验单如何解读?
7、肾上腺相关检查化验如何解读?
8、本患者17α羟化酶缺乏症的诊断充分吗?
9、地塞米松的作用?
10、HRT治疗的目的及方法?
2007年7月11日,第3次就诊,内分泌科、妇泌科
用药7+月。HRT后有月经来潮。
查体:身高160 cm,体重44 kg,血压120/80 mmHg,乳房Ⅳ级。
化验:ACTH:55.5 pg/ml↑。K:4.5 mmol/L。
处理:地塞米松减量,0.375 mg,1次/d,口服;克龄蒙1片,1次/d,口服;利维爱1.25 mg,1次/d,口服。定期体检。
2012年3月14日,复诊,22岁,妇泌科
用药5+年,追问患者近2年未体检,予全面检查。
ALT:179 U/L↑,AST:51 U/L↑,K:3.9 mmol/L。
盆腔超声:子宫4.2×3.7×2.5 cm,内膜1.0 cm,双附件区未见异常。
乳腺超声:双侧乳腺增生。
骨密度:T-3,骨质疏松。
处理:肝功异常,停用HRT相关药物,转内科予“美能”等保肝处理。
2012年至今,多次复诊
经保肝治疗后,ALT、AST正常。
继续原方案用药长期控制。嘱每年体检。
11、17α羟化酶缺乏症(46,XX)与(46,XY)临床处理有哪些不同?
2 解 析
(1)高血压、低血钾的常见原因有哪些?
常见原因包括:
① 盐皮质激素(醛固酮)增多:
原发性醛固酮增多症:如肾上腺醛固酮瘤。醛固酮的保水、保钠、排钾、排氢作用,临床表现为高血压及低血钾的相关症状。
继发性醛固酮增多症:肾素瘤、Bartter综合征、肾动脉狭窄、充血性心衰、肝硬化、大剂量利尿剂的应用等。
② 糖皮质激素分泌增多:Cushing综合征。
③ Liddle综合征:先天性肾小管遗传缺陷导致症状。
④ 先天性肾上腺皮质增生(CAH):17α羟化酶缺乏、11β羟化酶缺乏[1]。
(2)原发闭经的常见原因有哪些?
① 低促性腺激素:下丘脑垂体因素,最常见的是Kallmann综合征。
② 高促性腺激素:Turner综合征、卵巢不敏感综合征、46XX或46XY性腺发育不全、17α羟化酶缺乏、雄激素不敏感综合征等。
③ 正常促性腺激素:米勒管发育不全综合征、先天阴道闭锁、处女膜闭锁等。
(3)17α羟化酶缺乏症的发病机制?
17α羟化酶缺乏症为常染色体隐性遗传病,由CYPl7基因突变引起。17α羟化酶缺乏使孕烯醇酮不能转化为17α羟孕烯醇酮和17α羟孕酮,从而不能生成足够的性激素及皮质醇,反馈性地导致ACTH分泌过多引起双侧肾上腺皮质增生。同时因17a羟化过程受阻,孕酮等底物堆积,引起去氧皮质酮、醛固酮大量增加,促进保钠排钾,出现低血钾、血容量增多,血压升高,从而抑制了肾素-血管紧张素系统。这一系列的作用,导致患者可出现皮质醇、雌二醇、睾酮、肾素水平低下,双侧肾上腺增生或占位。
(4)17α羟化酶缺乏症有哪些特征及其原理?
常见主诉及临床表现:青春期无月经来潮、无第二性征发育、高血压;或青春期无月经来潮伴头痛乏力、软瘫抽搐、反复发现卵巢囊肿等。
超声:46,XX可见幼稚子宫,46,X提示子宫缺如,盆腔空虚。
查体:高血压、无阴毛腋毛,幼女型外阴,有阴道,46,XX有幼稚子宫,46,XY为阴道盲端。
电解质化验:低钾。
性激素检查:高促性腺激素、高P、低E2、低T、低17α-OHP。
内分泌化验:高ACTH、低皮质醇、低24h尿游离皮质醇、ACTH兴奋试验提示17α羟孕酮及血皮质醇水平无明显升高[2]。
(5)可疑17α羟化酶缺乏症需要作哪些临床检查?
盆腔超声、性激素六项、染色体、电解质、17α-OHP、肾上腺功能相关检查。
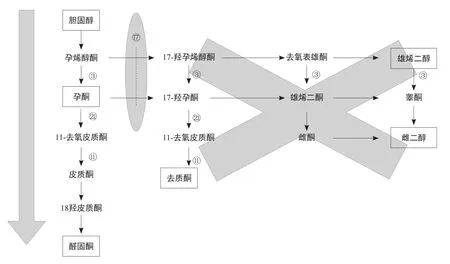
注:③ 3β-羟化脱氢酶11β羟化酶17α羟化酶21β羟化酶
(6)性激素化验单如何解读?
患者化验单提示高促性腺激素、低E2、低T。应充分鉴别高促性腺激素闭经相关疾病。若为垂体下丘脑低能抑制低下原因,包括P等也应一齐低下,而17α羟化酶缺乏疾病的一大特点是高P,是诊断的重要线索。此次检查未查P,较为遗憾,因此不可忽视孕酮的检查。
(7)肾上腺相关化验检查单如何解读?
肾上腺相关检查提示:醛固酮↑,ACTH↑,血总皮质醇↓,24小时尿游离皮质醇↓。
(8)本患者17α羟化酶缺乏症的诊断充分吗?
结合患者原发闭经、无第二性征发育、高血压低血钾、高促性腺激素、低E、低T,肾上腺相关检查提示:醛固酮试验↑,ACTH↑,血总皮质醇↓,24小时尿游离皮质醇↓,染色体46,XX。可诊断:完全型17α羟化酶缺乏症(46,XX)。
(9)地塞米松的作用?
糖皮质激素可补充肾上腺分泌皮质醇的不足,抑制ACTH过多释放,从而减少了去氧皮质酮等物质的分泌,抑制肾上腺增生,改善高血压和低血钾,还可以促进骨骺的闭合。
糖皮质激素的剂量根据患者体重、血压、血钾等的变化进行调整。国内报道应用地塞米松初始剂量为0.75~2 mg/d,维持剂量为0.1~0.375 mg/d,可使症状得到明显的改善,少数患者需加用降压药。
(10)HRT治疗的目的及方法?
青春期后的患者可以开始周期性服用雌激素以促进女性第二性征的发育,同时改善骨质疏松,避免骨折。
46,XY患者因无子宫,应用单雌即可;46,XX有子宫,应加用孕激素以保护内膜。
雌、雄激素合用对于提高腰椎和髋部骨密度的作用远远大于单用雌激素,因此本患者应用克龄蒙的同时加用了利维爱。
(11)17α羟化酶缺乏症(46,XX)与(46,XY)临床处理有哪些不同?
①HRT中,46,XY仅用单雌,46,XX需加用孕激素。
②46,XY患者性腺为睾丸,发育不良的异位睾丸恶变的发生率高,所以确诊后经补充适量的糖皮质激素至血压和血钾平稳时就可以手术切除以预防肿瘤发生。
3 病历完善
任××,社会性别女,17岁。2006年11月14日初次就诊。
主诉:原发闭经,发现高血压、低血钾3年。
现病史:本人为足月顺产而生,出生女性外阴,按女性社会性别抚养。自小生长发育及智力与常人无明显差别。近3年发现低血钾,最低1.35 mmol/L,间断静脉补钾及口服补钾。同时发现血压高,平时160/100 mmHg,最高血压170/140 mmHg。在当地医院住院治疗,可疑诊断为特发性醛固酮增多症或17α羟化酶缺乏症。至今无月经来潮,无第二性征自主发育。
既往史:否认特殊病史。
月经史:原发闭经。
个人史:生于当地,无不良嗜好。
家族史:有一妹,正常。否认家族中有类似病史。
查体:身高:160 cm,体重:41 kg;BP:160/100 mmHg,HR:60次/min;心肺未及异常,肝脾未及;乳腺Ⅰ~Ⅱ级(追问患者曾用过倍美力,具体不详);无腋毛;女性幼稚型外阴,无阴毛,有阴道;双下肢无水肿。
辅助检查:
染色体:46,XX。
盆腔超声:膀胱后方见子宫样结构,大小2.0×0.8×0.7 cm,肌层回声均,隐约可见内膜强回声;左附件区见卵巢样结构1.7×0.8 cm;右附件区件卵巢样结构1.8×0.9 cm。提示:幼稚子宫,卵巢发育不良?。
CT:双侧肾上腺增生。
肝肾功大致正常;K:2.90 mmol/L↓。
性激素:FSH:93.1 IU/L↑,LH:43.5 IU/L↑,E2:6.24 pmol/L↓,T:0 ng/mL↓。
卧立位醛固酮试验:Ald卧位9.0毫微克/L↑,立位10.0毫微克/L;PRA卧位0.2,立位0.2↓;AT-Ⅱ卧位124.1↑,立位115.2↑。
ACTH:95.2 pg/mL↑。
血总皮质醇:0.24 μg/dL↓。
24小时尿游离皮质醇(UFC):<0.20 μg/24hr↓。
诊断:完全型17α羟化酶缺乏症(46,XX)。
治疗经过:
地塞米松0.75 mg,1次/d,口服;克龄蒙1片,1次/d,口服;利维爱1.25 mg,1次/d,口服;钙片600 mg,1次/d,口服。3个月后复查B超。
HRT后有月经来潮。用药8个月后复查:BP:120/80 mmHg,乳房Ⅳ级,ACTH:55.5 pg/mL↑(较前好转),K:4.5 mmol/L。治疗方案:地塞米松减量,0.375 mg,1次/d,口服;克龄蒙1片,1次/d,口服;利维爱1.25 mg,1次/d,口服。
继续规律服药5年,复查发现:ALT:179 U/L↑,AST:51 U/L↑,K:3.9 mmol/L。盆腔超声:子宫4.2×3.7×2.5 cm,内膜1.0 cm,双附件区未见异常。乳腺超声:双侧乳腺增生。骨密度:骨质疏松。处理:停用HRT相关药物,转内科予“美能”等保肝处理。
经过保肝治疗,ALT、AST正常,继续原方案用药长期控制,嘱每年体检。
4 总 结
(1)17α羟化酶缺陷症(17-OHD)是先天性肾上腺皮质增生症(CAH)的少见类型,为常染色体隐性遗传病,由CYPl7基因突变引起。临床表现为原发闭经、性幼稚、高血压、低血钾、高促性腺激素、高P、低E低T、低17α羟孕酮等。按照染色体核型可分为46,XX和46,XY。其中46,XX更加少见。46,XX表现为原发性闭经、幼稚子宫、第二性征发育缺乏。46,XY表现为女性外阴和阴道盲端,腹腔睾丸,没有子宫、输卵管。一经确诊,需给予糖皮质激素、HRT治疗。HRT中,46,XY患者单用雌激素即可,46,XX患者需加用孕激素。46,XY患者性腺为睾丸,需手术切除以预防肿瘤发生。
(2)临床遇到闭经、性征发育障碍等妇科内分泌疾患可能的患者,应全面检查性激素六项,必要时需加查硫酸脱氢表雄酮、17α-OHP等指标,染色体是必要的检查项目不要遗漏。许多妇泌疾患涉及基因突变,可出现多系统异常表现,如生长迟滞、智力低下、电解质紊乱、糖脂代谢异常等,有时常常是先在其他科室就诊后转诊而来,因此,应加强多科室协作,加强多学科领域相关知识的学习。若有必要,请内分泌科、神经科等协助排除其他系统疾患。
(3)在疾病确诊后需要长期用药控制,应加强宣教及随访,注意定期复查肝肾功能、盆腔乳腺超声等,避免副反应。
参考文献
[1] 萧建中.高血压伴低血钾的诊断思维.中国实用内科杂志,2005,25(11):961-963.
[2] 王含必,田秦杰,孙爱军.完全型17α羟化酶缺乏48例临床分析.中华妇产科杂志,2012,47(7):518-521.
[3] 张 琳,王海宁,洪天配.17α羟化酶缺乏症一例的诊治经验及文献回顾.北京大学学报,2008,40(2):221-222.
本文编辑:张 钰
【中图分类号】R586
【文献标识码】A
【文章编号】ISSN.2095-8803.2016.05.016.04
