肿瘤坏死因子α通过PI3K-IP3R-Ca2+途径诱导乳鼠心肌肥大*
2016-07-05王桂君姚玉胜王洪新
王桂君, 姚玉胜, 王洪新
(辽宁医学院 1附属第一医院, 2附属第三医院, 3药理学教研室,辽宁 锦州 121000)
肿瘤坏死因子α通过PI3K-IP3R-Ca2+途径诱导乳鼠心肌肥大*
王桂君1,姚玉胜2△,王洪新3
(辽宁医学院1附属第一医院,2附属第三医院,3药理学教研室,辽宁 锦州 121000)
[摘要]目的: 探讨肿瘤坏死因子α(TNF-α)是否通过PI3K-IP3R-Ca2+信号途径诱导心肌肥大。方法: 以培养的乳鼠心肌细胞为模型,采用Lowry法测心肌细胞蛋白含量;[3H]-亮氨酸掺入法测定心肌细胞蛋白合成;计算机图像分析系统测心肌细胞体积;Till阳离子测定系统测定心肌细胞内Ca2+浓度。结果: PI3K阻断剂LY294002(50 μmol/L)明显抑制TNF-α(100 μg/L)诱导的心肌 [Ca2+]i增高(P<0.01),对正常心肌[Ca2+]i无明显影响;其抑制程度与合用2-氨基乙基二苯硼酸盐(2-APB)组相近,但小于与兰尼碱(RYA)合用组(P<0.05)。PI3K阻断剂LY294002(50 μmol/L)明显抑制TNF-α(100 μg/L)诱导的心肌细胞蛋白含量、蛋白合成及细胞体积的增加;其抑制程度与合用2-APB组无差异,但明显大于单用2-APB组,小于合用RYA组(P<0.05)。PI3K阻断剂LY294002(50 μmol/L)对正常心肌细胞蛋白含量、蛋白合成及细胞体积无明显影响。结论: TNF-α通过PI3K-IP3R-Ca2+途径诱导心肌肥大。
[关键词]心肌肥大; 肿瘤坏死因子α; PI3K-IP3R-Ca2+通路
心肌肥大的信号转导机制非常复杂,启动心肌肥大的信号转导通路一直是心血管疾病研究领域的重要问题。目前已了解到若干条细胞内信号通路包括磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)途径参与了心肌肥大的病理过程[1]。研究发现,PI3K-Akt/PKB途径的激活在肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)诱导培养乳鼠心肌细胞蛋白合成增加中起到重要作用[2],但对于其上、下游元素尚未确定。
近几年研究表明,PI3K参与心肌细胞Ca2+信号的调节[3]。有文献报道PI3K通过激活心肌细胞L型Ca2+通道增加心肌细胞内Ca2+水平,而与肌浆网钙泵[sarco(endo)plasmic reticulum calcium ATPase 2a,SERCA2a]、兰尼碱受体(ryanodine receptor,RyR)以及Na+/Ca2+交换体(Na+/Ca2+exchanger,NCX)无关[4]。也有研究表明心肌特定性PI3Kα过表达诱导Ca2+调节蛋白RyR、小窝蛋白(caveolin,Cav)1/2和SERCA2a的表达上调[5]。目前大量研究表明,PI3K的激活是磷脂酶C(phospholipase C,PLC)激活所必需的[6]。PI3K通过其产物磷脂酰肌醇-3,4,5-三磷酸(phosphatidylinositol 3,4,5-trisphosphate,PI-3,4,5-P3)激活PLC,PLC水解4,5二磷酸磷脂酰肌醇(phosphatidylinositol 4,5-bisphosphate,PI-4,5-P2)产生第二信使三磷酸肌醇(inositol 1,4,5-trisphosphate,IP3)和二酰甘油(diacylglycerol,DAG),诱导心肌细胞内Ca2+释放,增加心肌[Ca2+]i[7-8]。
心肌[Ca2+]i升高是导致心肌肥大的最基本信号,是心肌肥大的各种信号通路的汇集点,在心肌肥大的病理过程中起着重要作用。我们的研究[9-10]已证明TNF-α通过作用RyR和IP3R诱导心肌[Ca2+]i升高从而诱导心肌肥大。那么在TNF-α诱导心肌肥大过程中激活的PI3K是否参与了[Ca2+]i的调节?我们的研究[11]已表明TNF-α诱导心肌[Ca2+]i升高与L型Ca2+通道无关,故可以排除PI3K通过打开L型Ca2+通道参与调节TNF-α诱导的心肌[Ca2+]i升高的可能性。我们进一步探讨PI3K能否通过作用RyR或IP3R参与调节TNF-α诱导的心肌细胞内Ca2+浓度,从而诱导心肌肥大。
材料和方法
1动物
出生1~3 d的SD大鼠,雌雄兼用,由辽宁医学院实验动物中心提供。动物合格证号为SCXK(辽)20030007。
2主要试剂
低糖DMEM和胰蛋白酶(Gibco);5-溴脱氧尿嘧啶核苷(5-bromo-2′-deoxyuridine,BrdU)、维生素B12、转铁蛋白、SDS、牛血清白蛋白、IP3R阻断剂二苯硼酸2-氨基乙酯(2-aminoethoxydiphenyl borate,2-APB)和Fura-2/AM(Sigma);胎牛血清(杭州四季青生物材料有限公司); [3H]-亮氨酸(上海原子核研究所);TNF-α(R&D); RyR阻断剂兰尼碱(ryano-dine,RYA)购自Merck;PI3K阻断剂LY294002(Biosource)。
3主要方法
3.1体外乳鼠心肌细胞原代培养无菌条件下迅速取出心脏。Hanks液冲洗3次后剪成约1 mm×1 mm×1 mm大小碎块。加入0.08%胰蛋白酶消化细胞,将消化完毕细胞调至1×109/L(含84%DMEM培养基,15%胎牛血清、1×105U/L青霉素及100 mg/L链霉素)接种于24孔培养板,于5% CO2培养箱中培养。最初72 h用0.1 mmol/L BrdU抑制非心肌细胞生长。培养2~3 d换无血清DMEM培养基(内含5 mg/L胰岛素,5 μmol/L维生素B12,10 mg/L转铁蛋白)培养24 h。
3.2分组及给药心肌细胞培养分为7组:对照组;TNF-α组;2-APB+TNF-α组;LY294002+ TNF-α组;LY294002+2-APB+TNF-α组;LY294002+RYA+TNF-α组;LY294002组。2-APB、LY294002及RYA浓度分别为30、50、50 μmol/L,均在TNF-α(100 μg/L)加入前30 min加入。给药72 h后进行肥大指标包括心肌细胞蛋白含量、蛋白合成和细胞体积的测定。
3.3Fura-2/AM负载培养心肌细胞后采用Till阳离子测定系统测定心肌细胞[Ca2+]i瞬变共设9组:对照组;TNF-α组;2-APB+TNF-α组;RYA+TNF-α组;2-APB+RYA+TNF-α组;LY294002+TNF-α组;LY294002+2-APB+TNF-α组; LY294002+RYA+TNF-α组;LY294002组。上述阻断剂及TNF-α浓度同前所述。预孵育30 min后上机测定。先记录一段基础值,观察各阻断剂对正常心肌细胞[Ca2+]i瞬变有无影响;再加入TNF-α(100 μg/L),观察TNF-α刺激前后钙瞬变的变化。
3.4培养心肌细胞蛋白质含量的测定吸去培养板各孔中的培养液,用D-Hanks液快速冲洗3次后,加入1% SDS 0.5 mL溶解细胞,Lowrys法测每孔细胞蛋白质含量。
3.5培养心肌细胞蛋白质合成的测定将生长在24孔培养板上无血清静止培养了48 h的细胞培养液倒掉,代之以含有3.7×104Bq[3H]-leucine及各种浓度的试剂,另备一组无任何细胞的单纯培养基组,同样加入3.7×104Bq [3H]-leucine与其它组一起培养72 h。用1 mL 1%的SDS溶解细胞,用液闪仪测量[3H]-leucine的掺入量,进行蛋白合成的分析。
3.6培养心肌细胞体积的测定收集消化下的细胞注入一细胞室内,在放大400倍的倒置显微镜下观察细胞,几乎均呈球形,用计算机CIAS大恒细胞图象分析系统测量单个细胞的直径,进而计算出细胞体积。每孔随机选择4个视野,每个视野测20个细胞。
3.7培养心肌细胞[Ca2+]i瞬间变化的测定将长有自发性搏动的心肌细胞的盖玻片从培养皿中取出,置于含有Fura-2/AM(3 μmol/L)的DMEM培养基中,其中含有白蛋白0.2%,在37 ℃水浴中孵育30 min。取出盖玻片,用HEPES缓冲液冲洗后,放于荧光显微镜下的灌流槽中,恒温37 ℃,用HEPES缓冲液灌流,灌流速度为1 mL/min,所有药物均在指定时间加入灌流液中。所用的测定仪器为Till阳离子测定系统。激发光波长分别为340及380 nm,发射光波长为505 nm,采样间隙为300 ms。每次选取5~10个细胞测量心肌[Ca2+]i的瞬变,连续记录给药前后的荧光强度。根据文献[12]方法计算心肌细胞[Ca2+]i,计算前应减去细胞自身的荧光。
4统计学处理
采用SPSS 13.0统计软件进行数据统计。数据均以均数±标准差(mean±SD)表示,采用单因素方差分析和析因设计的方差分析进行统计学检验,各组间均数间的两两比较采用SNK-q检验。以P<0.05为差异有统计学意义。
结果
1PI3K对TNF-α诱导的心肌细胞内钙离子瞬变的影响
PI3K阻断剂LY294002并不明显改变正常培养心肌细胞的 [Ca2+]i瞬变幅度、静息水平及搏动频率。LY294002明显抑制由TNF-α诱导的心肌细胞内钙离子瞬变幅度增高,但不影响基线水平及心肌细胞自发搏动频率。其抑制程度与2-APB或RYA的抑制作用相近,但较二者合用作用小(P<0.01)。LY294002与2-APB合用组对TNF-α诱导的心肌细胞内Ca2+瞬变增高的抑制作用与LY294002单用组差异无统计学意义,提示TNF-α激活PI3K诱导的心肌细胞内Ca2+瞬变增高与IP3R有关。LY294002与RYA合用组对TNF-α诱导的心肌细胞内Ca2+瞬变增高的抑制作用明显大于LY294002单用组(P<0.01),提示TNF-α激活PI3K诱导的心肌细胞Ca2+瞬变升高与RyR无关,见图1。

Figure 1.The effects of PI3K on the spontaneous [Ca2+]itransient and rest/peak amplitude in cultured ventricular myocytes from the neonatal rats treated with TNF-α. LY: LY294002. Mean±SD.n=4.**P<0.01vscontrol;##P<0.01vsTNF-α;△△P<0.01vsLY+TNF-α.
图1PI3K对TNF-α诱导的心肌细胞内钙离子瞬变的幅度、静息值和频率的影响
2LY294002在有或无2-APB/RYA存在时对TNF-α诱导的心肌细胞蛋白含量增加的影响
与对照组相比,TNF-α组的心肌细胞蛋白含量增加了48.87%(P<0.01);LY294002组的心肌细胞蛋白含量未见明显改变,说明LY294002对正常心肌细胞蛋白含量无明显影响。与TNF-α组相比,TNF-α+2-APB组、TNF-α+LY294002组和TNF-α+LY294002+2-APB组的心肌细胞蛋白含量明显降低(P<0.01),分别降低了19.29%、33.47%和35.26%,说明TNF-α通过激活PI3K或IP3R诱导心肌细胞蛋白含量增加。其抑制程度LY294002组与LY294002+2-APB组接近,但明显大于2-APB组(P<0.05);提示TNF-α激活的PI3K通过作用IP3R诱导心肌细胞蛋白含量增加,但除此可能尚存在其它途径。LY294002+RYA组显著抑制TNF-α诱导的心肌细胞蛋白含量增加(P<0.01),降低了49%,与TNF-α+LY294002组相比有显著差异(P<0.05),说明TNF-α激活的PI3K不是通过作用于RyR来参与诱导心肌蛋白含量增加的,见图2。
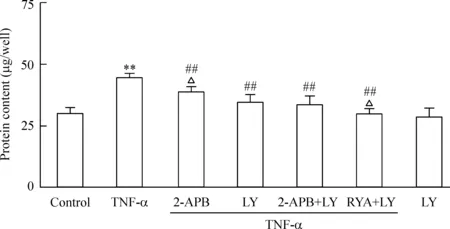
Figure 2.The effects of LY294002, RYA and/or 2-APB on protein content of cultured ventricular myocytes from neonatal rats treated with TNF-α. LY: LY294002. Mean±SD.n=8.**P<0.01vscontrol;##P<0.01vsTNF-α;△P<0.05vsLY+TNF-α.
图2LY294002在有或无2-APB/RYA时对TNF-α诱导的心肌细胞蛋白含量增加的影响
3LY294002在有或无2-APB/RYA存在时对TNF-α诱导的心肌细胞蛋白合成增加的影响
与对照组相比,TNF-α组的心肌细胞蛋白合成增加了33.97%(P<0.01);LY294002组的心肌细胞蛋白合成量未见明显改变,说明LY294002对正常心肌细胞蛋白合成无明显影响。与TNF-α组相比,TNF-α+2-APB组、TNF-α+LY294002组、TNF-α+LY294002+2-APB组的心肌细胞蛋白合成明显降低(P<0.01),分别降低了13.31%、23.40%和24.05%,说明TNF-α通过激活PI3K或IP3R诱导心肌细胞蛋白合成增加;其抑制作用LY294002组与LY294002+2-APB组接近,但明显大于2-APB组(P<0.05),说明PI3K通过作用IP3R调节TNF-α诱导的心肌细胞蛋白合成增加,但除此PI3K可能还存在其它途径参与TNF-α诱导的心肌蛋白合成增加。TNF-α+LY294002+ RYA组显著抑制TNF-α诱导的心肌细胞蛋白合成增加(P<0.01),降低了约32.98%,与TNF-α+LY294002组相比差异有统计学显著性(P<0.05),说明PI3K不是通过作用于RyR来调节TNF-α诱导的心肌蛋白合成增加的,见图3。
4LY294002在有或无2-APB/RYA存在时对TNF-α诱导的心肌细胞体积增加的影响
与对照组相比,TNF-α组的心肌细胞体积增加了47.57% (P<0.01);LY294002组的心肌细胞体积未见明显改变,说明LY294002对正常心肌细胞体积无明显影响。与TNF-α组相比,TNF-α+ 2-APB组、TNF-α+LY294002组和TNF-α+ LY294002+2-APB组的心肌细胞体积明显降低(P<0.01),分别降低了13.94%、25.06%和31.51%,说明TNF-α通过激活PI3K或IP3R诱导心肌细胞体积增加。LY294002的抑制作用与LY294002+2-APB合用组接近,但显著大于单用2-APB组(P<0.01),说明PI3K通过作用IP3R调节TNF-α诱导的心肌细胞体积增大,但除此尚存在其它途径。LY294002+RYA组显著抑制TNF-α诱导的心肌细胞体积增大(P<0.01),降低了约40.33%,与TNF-α+LY294002组相比有显著差异(P<0.01),说明PI3K不是通过作用于RyR来调节TNF-α诱导的心肌细胞体积增大的,见图4。

Figure 3.The effects of LY294002, RYA and/or 2-APB on [3H]-leucine incorporation of cultured ventricular myocytes from neonatal rats treated with TNF-α. LY: LY294002. Mean±SD.n=8.**P<0.01vscontrol;##P<0.01vsTNF-α;△P<0.05vsLY+TNF-α.
图3LY294002在有或无2-APB/RYA存在时对TNF-α诱导的心肌细胞蛋白合成增加的影响

Figure 4.The effects of LY294002, RYA and/or 2-APB on the cell size of cultured ventricular myocytes from neonatal rats treated with TNF-α. LY: LY294002. Mean±SD.n=80.**P<0.01vscontrol;##P<0.01vsTNF-α;△△P<0.01vsLY+TNF-α.
图4LY294002在有或无2-APB/RYA存在时对TNF-α诱导的心肌细胞体积增加的影响
讨论
心肌肥大是心脏对多种因素长期刺激所发生的适应性改变,早期的代偿性心肌肥大和心功能适应性改变对机体是有利的,但若长期得不到纠正将逐渐演变为心力衰竭[13]。由心衰而死亡是临床病人的主要死因之一。现在研究认为,在心肌肥大发生发展过程中,胞内钙信号途径是最重要信号转导通路之一。
已经发现PI3K抑制剂有LY294002和wortmannin两种。我们之所以选择LY294002,主要是因为与wortmanni相比,LY294002对PI3K的选择性要更专一[14]。本研究显示,PI3K阻断剂LY294002(50 μmol/L)显著抑制TNF-α诱导的心肌内钙离子瞬变幅度增高,但不影响基线水平及心肌细胞自发搏动频率。我们之前的研究[15]已证明L型Ca2+通道与TNF-α诱导的幅度增高无关,故可以排除PI3K通过打开L型Ca2+通道促使TNF-α诱导心肌细胞内钙离子瞬变幅度增高的可能。基于我们先前的研究[9, 16]:TNF-α诱导心肌细胞内钙离子瞬变幅度增高与IP3R和RyR有关,我们进一步观察在用IP3R阻断剂2-APB(30 μmol/L)或RyR阻断剂RYA(50 μmol/L)分别阻断IP3R或RyR后LY294002对TNF-α诱导的心肌细胞内钙离子瞬变的影响。结果发现PI3K阻断剂LY294002明显抑制了TNF-α诱导的[Ca2+]i升高,提示PI3K激活参与调节TNF-α诱导的心肌细胞[Ca2+]i升高。LY294002与2-APB合用组对TNF-α诱导的心肌细胞内Ca2+瞬变增高的抑制作用与LY294002单用组比较差异无统计学意义,提示TNF-α激活PI3K诱导的心肌细胞内Ca2+瞬变增高与IP3R有关。LY294002与RYA合用组对TNF-α诱导的心肌细胞内Ca2+瞬变增高的抑制作用明显大于LY294002单用组,提示TNF-α激活PI3K诱导的心肌细胞Ca2+瞬变升高与RyR无关。
为了明确[Ca2+]i升高在TNF-α诱导心肌肥大PI3K通路中的作用,我们通过检测心肌细胞蛋白含量、蛋白合成量以及心肌细胞体积等指标,进一步观察在有或无2-APB、RYA存在时LY294002对TNF-α诱导的心肌细胞肥大的影响。结果表明PI3K阻断剂LY294002显著抑制TNF-α诱导的心肌细胞蛋白含量、蛋白合成量以及细胞体积的增加,其抑制程度与LY294002+2-APB无显著差异。基于我们先前的研究已经表明2-APB对TNF-α诱导的心肌肥大的抑制作用是由于2-APB阻断IP3R导致TNF-α诱导的[Ca2+]i升高受到抑制,故目前研究提示TNF-α激活的PI3K可能是通过作用IP3R引起心肌细胞[Ca2+]i增加,从而诱导心肌细胞肥大的。但LY294002的抑制程度明显大于2-APB,提示TNF-α激活的PI3K不完全通过调节[Ca2+]i参与诱导肥大,除此之外可能尚存在其它途径。LY294002与RYA合用对TNF-α诱导心肌肥大的抑制作用与LY294002单用组相比有显著差异,说明PI3K不是通过作用于RyR调节[Ca2+]i,从而参与TNF-α诱导心肌肥大的。这与我们前面结果所表明的PI3K通过作用IP3R而非RyR调节TNF-α诱导的心肌[Ca2+]i增加相一致,进一步支持[Ca2+]i增加在TNF-α诱导心肌肥大PI3K通路中起作用。
本研究结果说明TNF-α通过PI3K-IP3R-Ca2+途径诱导心肌肥大。TNF-α激活PI3K诱导的心肌肥大仅部分通过IP3R-Ca2+信号途径,除此可能尚存在其它途径参与TNF-α诱导的心肌肥大。据此可以推测,在TNF-α诱导心肌肥大的PI3K通路中,Ca2+信号通路与先前文献报道的PI3K-Akt/PKB途径不是一路,至少在Ca2+水平上是这样。至于其后两路是否会有交叉或交互,尚待进一步研究。
[参考文献]
[1]Selvetella G, Hirsch E, Notte A, et al. Adaptive and maladaptive hypertrophic pathways: points of convergence and divergence[J]. Cardiovasc Res, 2004, 63(3):373-380.
[2]Hiraoka E, Kawashima S, Takahashi T, et al. TNF-α induces protein synthesis through PI3-kinase-Akt/PKB pathway in cardiac myocytes[J]. Am J Physiol Heart Circ Physiol, 2001, 280(4):H1861-H1868.
[3]董家龙,李菊香,孙国芳,等. Rock可能通过抑制PI3K/Akt通路促进缺氧诱导的心肌细胞凋亡[J].中国病理生理杂志,2014, 30(11):2071-2075.
[4]McDowell SA, McCall E, Matter WF, et al. Phosphoinositide 3-kinase regulates excitation-contraction coupling in neonatal cardiomyocytes[J]. Am J Physiol Heart Circ Physiol, 2004, 286(2):H796-H805.
[5]Yano N, Tseng A, Zhao TC, et al. Temporally controlled overexpression of cardiac-specific PI3Kα induces enhanced myocardial contractility: a new transgenic model[J]. Am J Physiol Heart Circ Physiol, 2008, 295(4):H1690-H1694.
[6]Maffucci T, Raimondi C, Abu-Hayyeh S, et al. A phosphoinositide 3-kinase/phospholipase Cγ1 pathway regulates fibroblast growth factor-induced capillary tube formation[J]. PLoS One, 2009, 4(12):e8285.
[7]Rameh LE, Rhee SG, Spokes K, et al. Phosphoinositide 3-kinase regulates phospholipase Cγ-mediated calcium signaling[J]. J Biol Chem, 1998, 273(37):23750-23757.
[8]Xie Z, Chang SM, Pennypacker SD, et al. Phosphatidylino-sitol-4-phosphate 5-kinase 1α mediates extracellular calcium-induced keratinocyte differentiation[J]. Mol Biol Cell, 2009, 20(6):1695-1704.
[9]王桂君,郭莲怡,王洪新. 钙离子在肿瘤坏死因子α诱导心肌肥大中的作用[J].中国动脉硬化杂志,2013, 21(1):47-51.
[10]王桂君,姚玉胜,王洪新. Ca2+/CaMKⅡ信号通路在肿瘤坏死因子-α诱导心肌肥大中的作用[J].中国药理学通报,2010, 26(3):387-391.
[11]王桂君,姚玉胜,李戆鹏,等. Ca2+信号在肿瘤坏死因子-α诱导心肌肥大PI3-K信号途径中的作用[J].中国应用生理学杂志,2010,26(3):284-288.
[12]Passier R, Zeng H, Frey N, et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factorinvivo[J]. J Clin Invest, 2000, 105(10):1395-1406.
[13]邹剑,周后凤,先志伟,等. 激活PPARα表达对AngⅡ诱导的心肌细胞肥大及NFATc4与p65-NFκB相互作用的影响[J]. 中国病理生理杂志, 2014, 30(6):1017-1022.
[14]Morita M, Yoshizaki K, Nakane A, et al. Inhibitory effect of the phosphoinositide 3-kinase inhibitor LY294002 on muscarinic acetylcholine receptor-induced calcium entry in PC12h cells[J]. J Pharmacol Sci, 2007, 105(3):258-263.
[15]王桂君,姚玉胜,李戆鹏,等. 肿瘤坏死因子α诱导心肌肥大中Ca2+升高与L型钙通道无关[J]. 中华高血压杂志,2010, 18(7):671-676.
[16]Wang GJ, Wang HX, Yao YS, et al. The role of Ca2+/calmodulin-dependent protein kinase II and calcineurin in TNF-α-induced myocardial hypertrophy[J]. Braz J Med Biol Res, 2012, 45(11):1045-1051.
(责任编辑: 卢萍, 罗森)
Neonatal rat cardiomyocyte hypertrophy is induced by tumor necrosis factor-α through PI3K-IP3R-Ca2+pathways
WANG Gui-jun1, YAO Yu-sheng2, WANG Hong-xin3
(1TheFirstAffiliatedHospital,2TheThirdAffiliatedHospital,3DepartmentofPharmacology,LiaoningMedicalCollege,Jinzhou121000,China.E-mail:wgj-april@163.com)
[ABSTRACT]AIM: To investigate the role of PI3K-IP3R-Ca2+pathways in cardiomyocyte hypertrophy induced by tumor necrosis factor-α (TNF-α). METHODS: Myocardial cells of neonatal rats were cultured in vitro. The hypertrophic model was induced by TNF-α. The protein content was assayed with Lowry’s method. The volumes of the cardiomyocytes were detected by computer photograph analysis system. The protein synthesis was determined by the method of [3H]-leucine incorporation. [Ca2+]i transient was measured by Till image system with cell-loading Fura-2/AM. RESULTS: LY294002, a PI3K inhibitor, significantly suppressed the amplitude elevation of the spontaneous [Ca2+]i transients induced by TNF-α in cultured ventricular myocytes from neonatal rats. The effect was similar to that of LY294002+2-APB (P>0.05), but lower than that in LY294002+ryanodine group (P<0.05). LY294002 significantly reduced the enhancements of protein content, [3H]-leucine incorporation and cell size induced by TNF-α. The effect was similar to that in 2-APB+LY294002 group, but higher than that in 2-APB group and lower than that in ryanodine+LY294002 group. CONCLUSION: TNF-α induces cardiac hypertrophy through PI3K-IP3R-Ca2+pathways.
[KEY WORDS]Cardiomyocyte hypertrophy; Tumor necrosis factor-α; PI3K-IP3R-Ca2+pathway
[文章编号]1000- 4718(2016)01- 0021- 06
[收稿日期]2015- 05- 20[修回日期] 2015- 10- 23
*[基金项目]辽宁省科技厅计划(No. 2013225305);辽宁医学院校长基金资助项目(No. xzjj20130232)
通讯作者△Tel: 0416-4197472; E-mail: wgj-april@163.com
[中图分类号]R363
[文献标志码]A
doi:10.3969/j.issn.1000- 4718.2016.01.004
